

70-07 節錄
The principal function of high-density lipoprotein (HDL) is to facilitate the reverse cholesterol transport (RCT) and inhibition of atheroma forma- tion. Epidemiological studies and interventional trials have suggested that HDL has cardioprotective properties.
However, increasing HDL concentra- tion may not necessarily increase RCT, especially if the increase in HDL levels is the result of inhibiting HDL cholesterol (HDL-C) flux. The results of recent phase III clinical trials utilizing a cholesterol ester transfer protein (CETP) inhibitor to increase HDL-C levels in hypoalphalipoproteinaemia have shown that this approach of elevating HDL-C levels is insufficient to combat atherosclerosis and reduce the risk of cardiovascular disease. Although there are several interventions that increase HDL-C by preventing its turnover in the circulation, a more desirable approach is to enhance de novo production of HDL in the liver and/or small intestine. To this end, our acquired knowledge of the apolipoprotein-I (apo A-I) gene promoter as well as the signalling pathways that modulate its expression, has fuelled the development of novel therapeutic strategies to increase HDL-C flux. Pro- mising pharmacological agents that selectively regulate transcription of the apo A-I gene, therapeutic strategies to de-repress apo A-I gene expression, and infusion of recombinant apo A-I or apo A-I mimetics are under devel- opment and may be clinically beneficial in the near future.
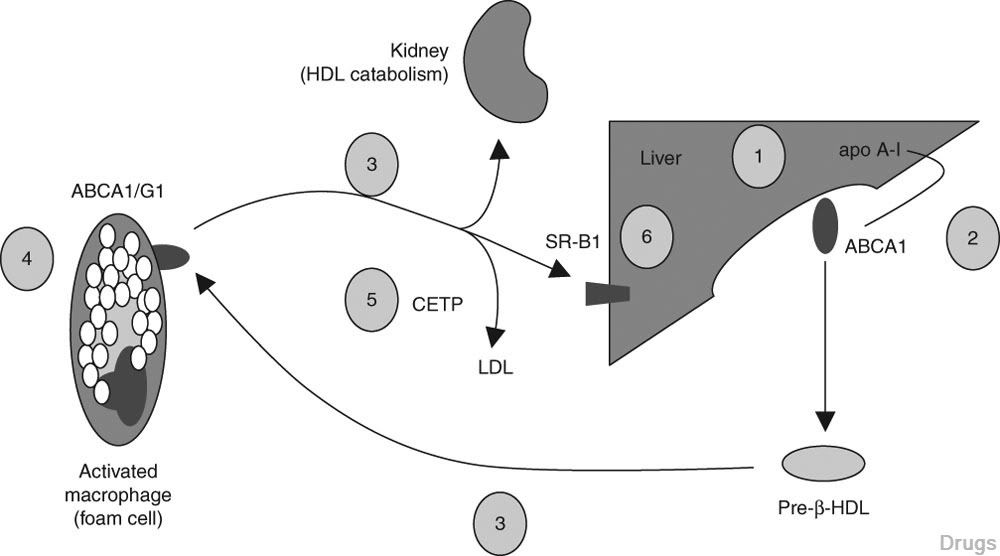
Fig. 1. Major pathways for development of high-density lipoprotein (HDL)-related therapies. (1) Increased synthesis of apolipoprotein A-I (apo A-I); (2) increased hepatic adenosine triphosphate-binding cassette protein A1 (ABCA1) synthesis; (3) preservation of HDL function (antioxidant properties, immunomodulatory functions, lipid acceptor properties); (4) increased ABCA1/G1 synthesis in HDL-target tissues (e.g. activated macrophage cells or ‘foam cells’); (5) prolongation of HDL half-life and increased recycling; and (6) hepatic cholesterol storage and bile acid metabolism. CETP = cholesterol ester transfer protein; LDL = low-density lipoprotein; SR-B1 = scavenger receptor B1.
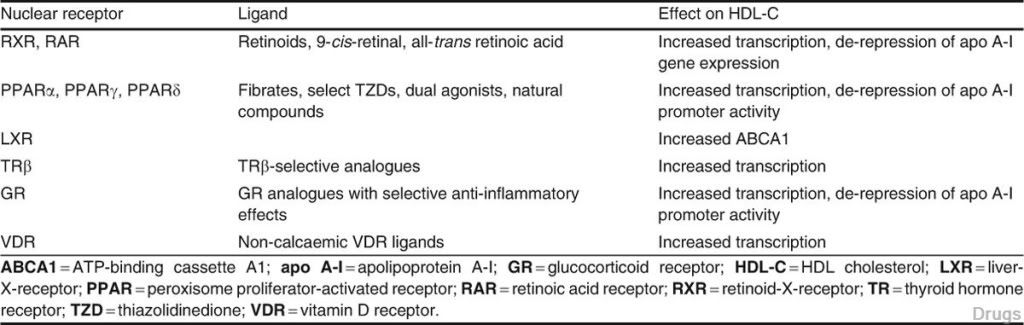
Table I. Targeting high-densitylipoprotein (HDL) by nuclear receptor agonists/antagonists
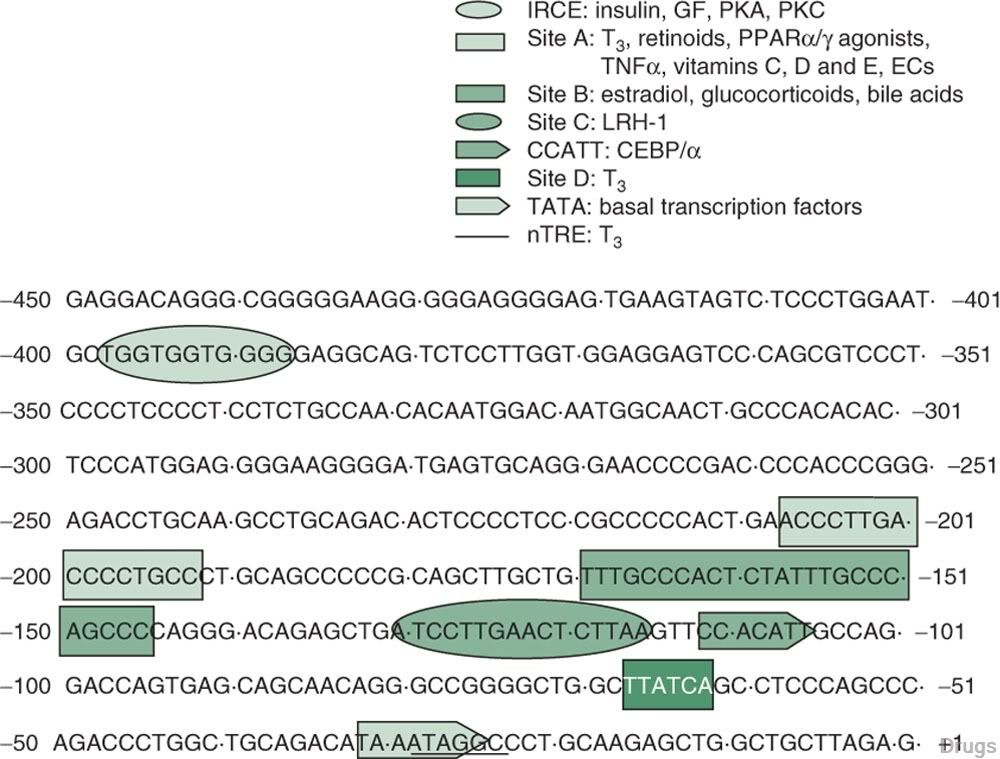
Fig. 2. Regions of the human apolipoprotein A-I (apo A-I) gene promoter that respond to various drug, metabolic, nutrient and growth signalling pathways. Sites A, B, C and D refer to regions of the apo A-I gene promoter that are protected from DNase I digestion in footprinting experiments. The conserved CCATT and TATA boxes bind the hepatocyte-enriched transcription factor CAT-box element binding protein-a and the basal transcriptional apparatus, respectively. Growth factors (GFs) and protein kinase A (PKA) and protein kinase C (PKC) modulate apo A-I promoter activity through the insulin-responsive core element (IRCE). Thyroid hormone (T3), retinoids, peroxisome proliferator- activated receptor-a and -g (PPARa/g) agonists, tumour necrosis factor-a (TNFa), vitamins C, D and E, and endocannabinoids (ECs) regulate apo A-I promoter activity through site A. Estradiol, glucocorticoids and bile acids signal require site B, and the orphan nuclear receptor liver receptor-homologue-1 (LRH-1) binds site C. Site D and the negative thyroid hormone responsive element (nTRE) are responsive to T3. However, whereas site D confers positive effects of T3 on apo A-I promoter activity, the nTRE inhibits apo A-I promoter activity in a T3-dependent manner.
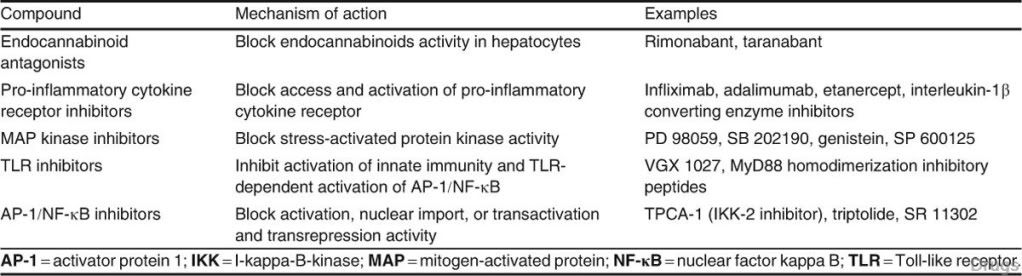
Table II. Targeting high-density lipoprotein (HDL) by de-repressing apolipoprotein A-I (apo A-I) gene expression


 留言列表
留言列表
 線上藥物查詢
線上藥物查詢 