

January 1, 2014DOI: 10.1056/NEJMoa1305224
Osteoporosis is characterized by low bone mass and defects in microarchitecture that are responsible for decreased bone strength and increased risk of fracture.1 Antiresorptive drugs for osteoporosis increase bone mineral density and prevent the progression of structural damage but may not restore bone structure. Stimulation of bone formation is necessary to achieve improvements in bone mass, architecture, and strength.
Sclerostin, encoded by the gene SOST, is an osteocyte-secreted glycoprotein that has been identified as a pivotal regulator of bone formation. By inhibiting the Wnt and bone morphogenetic protein signaling pathways, sclerostin impedes osteoblast proliferation and function, thereby decreasing bone formation.2-4 The fact that SOST expression is limited to skeletal tissue makes the inhibition of sclerostin a particularly attractive target, because it would affect skeletal health but limit the risk of off-target effects.5
Studies of the molecular effects of sclerostin support the concept that blocking the action of sclerostin results in positive effects on the skeleton. Patients with a genetic deficiency of sclerostin have high bone mass, correspondingly increased bone strength, and resistance to fractures.6,7 Mice in which the sclerostin gene was deleted had increased bone formation and high bone mass and strength.8 In estrogen-deficient rat and monkey models of postmenopausal osteoporosis, treatment with antisclerostin antibodies restored bone mass and bone strength to levels higher than those in control animals.9,10 Furthermore, Wnt activation resulting from the inhibition of sclerostin has been associated with decreased bone resorption both in humans and in animal models, probably owing to direct or indirect actions on osteoclasts through the Wnt pathway.11,12
Romosozumab (formerly known as AMG 785/CDP7851, Amgen and UCB Pharma) is a humanized monoclonal anti-sclerostin antibody. In a phase 1 study, single injections of romosozumab stimulated bone formation, decreased bone resorption, and increased bone mineral density.13 Here we report the results of a 1-year, phase 2 study evaluating the efficacy and safety of romosozumab in postmenopausal women with low bone mass.
METHODS
Study Design
We conducted a phase 2, multicenter, international, randomized, placebo-controlled, parallel-group, eight-group study, in which the primary end point was the percentage change from baseline in bone mineral density at the lumbar spine at 12 months. We enrolled 419 participants at 28 study centers in Argentina, Austria, Belgium, Canada, Denmark, Spain, and the United States. A total of 367 of the participants were randomly assigned to one of five dosing regimens of subcutaneous romosozumab (70 mg, 140 mg, or 210 mg once monthly, or 140 mg or 210 mg every 3 months) or to one of two open-label comparators (70 mg of oral alendronate weekly or 20 μg of subcutaneous teriparatide daily) (Figure 1
FIGURE 1
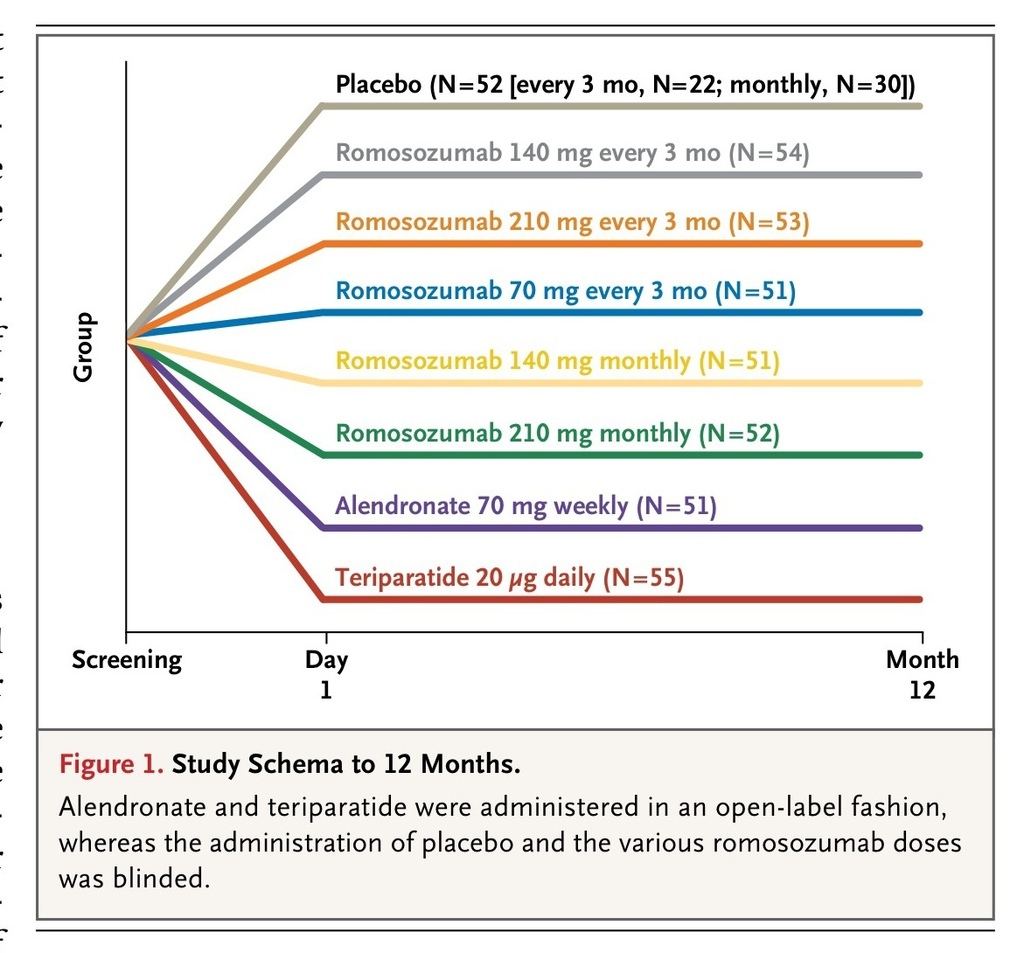
Study Schema to 12 Months.
). The remaining 52 participants were randomly assigned to a group that received placebo injections either monthly or every 3 months.
The randomization was performed by means of an interactive voice-response system according to a schedule prepared by the sponsor before initiation of the study. In the romosozumab and placebo groups, participants, study-site investigators, and study personnel were unaware of the study drug (romosozumab or placebo) and dose, but were aware of the dosing schedule (once monthly or every 3 months). Placebo groups received doses at the same frequency as the counterpart romosozumab groups. Throughout the study, all the participants were required to take at least 1000 mg of elemental calcium and 800 IU of vitamin D daily.
Study Oversight
The study protocol, available with the full text of this article at NEJM.org, was approved by the ethics committee or institutional review board at each center. Each participant provided written informed consent.
Representatives of Amgen designed the study in collaboration with some of the study investigators and UCB Pharma and performed the analysis according to a prespecified statistical analysis plan. The first author and two authors who are employees of Amgen take primary responsibility for the data and analyses and for the fidelity of the study to the protocol. The first author wrote the first draft of the manuscript with editorial support from Amgen. All the authors contributed to subsequent drafts of the manuscript and made the decision to submit the manuscript for publication. Clinical-trial agreements between Amgen and the investigators included provisions relating to confidentiality of the study data, a practice that is standard for clinical trials in the development phase of new pharmaceutical agents.
Study Participants
Ambulatory postmenopausal women, 55 to 85 years of age, were eligible if they had low bone mineral density (a T score of −2.0 or less at the lumbar spine, total hip, or femoral neck and −3.5 or more at each of the three sites). Exclusion criteria were a history of vertebral fracture or a fragility fracture of the wrist, humerus, hip, or pelvis after 50 years of age; a history of metabolic bone disease; a serum level of 25-hydroxyvitamin D of less than 20 ng per milliliter; untreated hyperthyroidism or hypothyroidism; current hyperparathyroidism or hypoparathyroidism; an elevated aminotransferase level; substantially impaired renal function (estimated creatinine clearance, ≤30 ml per minute, as assessed by means of the Modification of Diet in Renal Disease equation14); current hypercalcemia or hypocalcemia; cancer; a positive test for the human immunodeficiency virus, hepatitis C virus, or hepatitis B surface antigen; and a history of spinal stenosis, facial-nerve paralysis, or solid-organ or bone marrow transplantation. In addition, the use of any of the following agents affecting bone metabolism was an exclusion criterion: intravenous bisphosphonate or denosumab at any time; fluoride (for treatment of osteoporosis) within the previous 24 months; oral bisphosphonate, parathyroid hormone, or strontium within the previous 12 months; calcitonin, selective estrogen-receptor modulator, systemic oral or transdermal estrogen, or tibolone within the previous 3 months; or systemic glucocorticoid (≥5 mg of prednisone equivalent per day for >10 days) within the previous 3 months.
Study Procedures
Bone mineral density was measured at the lumbar spine and proximal femur by means of dual-energy x-ray absorptiometry (GE Healthcare or Hologic) at baseline and months 3, 6, and 12 and at the distal third of the radius at baseline and month 12. Scanning of the lumbar spine and proximal femur was performed in duplicate at baseline and month 12. Analysis of the scans and quality control of the scanners and the individual scans were provided by Synarc.
We assessed levels of the bone-formation markers procollagen type I N-terminal propeptide (UniQ P1NP RIA, Orion Diagnostica), osteocalcin (1-49 and peptide 1-43; OSTEO-RIACT kit, Cisbio Bioassays), and bone-specific alkaline phosphatase (Access Ostase, Beckman Coulter) and the bone-resorption marker β-isomer of the C-terminal telopeptide of type I collagen (β-CTX)(Serum CrossLaps ELISA, Nordic Bioscience Diagnostics) in fasting serum samples at baseline, week 1, and months 1, 2, 3, 6, 9, and 12 at the University of Liege. Levels of calcium and intact parathyroid hormone, chemical and hematologic values, and serum levels of anti-romosozumab antibodies were assessed at baseline and months 1, 3, 6, 9, and 12. Antibody development was evaluated with the use of a validated electrochemiluminescent immunoassay, and positive samples were tested for romosozumab-neutralizing activity in vitro. A detailed description of the antibody assays is provided in the Supplementary Appendix, available at NEJM.org.
Adverse events were reported spontaneously and in response to nondirected questioning at each study visit. The small number of participants in each group precluded an assessment of the effect of therapy on the incidence of fracture.
Primary and Secondary End Points
The primary end point was the percentage change from baseline in bone mineral density at the lumbar spine at month 12 in the individual romosozumab groups and the pooled placebo group. Key secondary end points included the percentage change from baseline in bone mineral density at the lumbar spine at month 6, at the total hip and femoral neck at months 6 and 12, and at the distal third of the radius at month 12, and the percentage change from baseline in the measures of bone metabolism at months 1, 3, 6, 9, and 12 in the individual romosozumab groups and pooled placebo group. Key exploratory end points included the effect of romosozumab as compared with alendronate or teriparatide on the percentage change from baseline in bone mineral density at the lumbar spine, total hip, femoral neck, and distal third of the radius over the 12-month period.
Statistical Analysis
The sample size was calculated on the basis of the comparison between romosozumab and placebo of the percentage change from baseline to month 12 in bone mineral density at the lumbar spine and total hip. We assumed that the mean (±SD) percentage changes in bone mineral density at the lumbar spine would be 0±3.6 in the placebo group and 5.0±3.6 in the romosozumab group and that the corresponding changes at the total hip would be 0±2.6 in the placebo group and 2.0±2.6 in the romosozumab group. Allowing for an average withdrawal rate of 15% across treatment groups in the first 12 months and a two-sided type I error of 5%, we calculated that a sample of 50 participants per group would provide the study with more than 90% power to detect those changes in bone mineral density at the lumbar spine and total hip, with the use of a two-sample t-test.
A linear mixed-effects model was fit with the percentage change from baseline to months 3, 6, and 12 in bone mineral density at the lumbar spine as the dependent variable and with baseline bone mineral density, machine type, interaction of baseline bone mineral density with machine type, geographic region, visit, treatment regimen (categorical), and interaction of treatment regimen with visit as the independent variables. The effect of the dose frequency of romosozumab (monthly vs. every 3 months) was estimated by means of pooling the dose groups within each frequency and comparing them with placebo. Similarly, the effect of the two highest doses of romosozumab (140 mg vs. 210 mg) was assessed by means of pooling the frequencies and comparing them with placebo. Hochberg's method was used to control for multiplicity of these four comparisons. Differences between each romosozumab regimen and the pooled placebo group, alendronate group, or teriparatide group were also estimated with the use of pair-wise contrasts at month 12. Analyses of the percentage change from baseline in bone mineral density and bone-turnover markers included all participants who had undergone randomization, had a nonmissing baseline value, and had at least one measurement after the baseline visit.
Analyses of safety included all participants who underwent randomization and received at least one dose of a study drug. Safety end points included the incidence and severity of adverse events; changes from baseline in vital signs, laboratory values, and electrocardiographic variables; and the incidence of the formation of anti-romosozumab antibodies.
RESULTS
Participants
A total of 419 participants were enrolled in the study and underwent randomization, and 383 (91%) completed the 12-month visit; 36 participants (9%) withdrew from the study (Fig. S1 in the Supplementary Appendix). The main reason reported for study discontinuation was withdrawal of consent (5% of participants). The demographic characteristics and key characteristics at baseline among the women enrolled in the study were balanced across the eight randomized groups (Table 1
TABLE 1
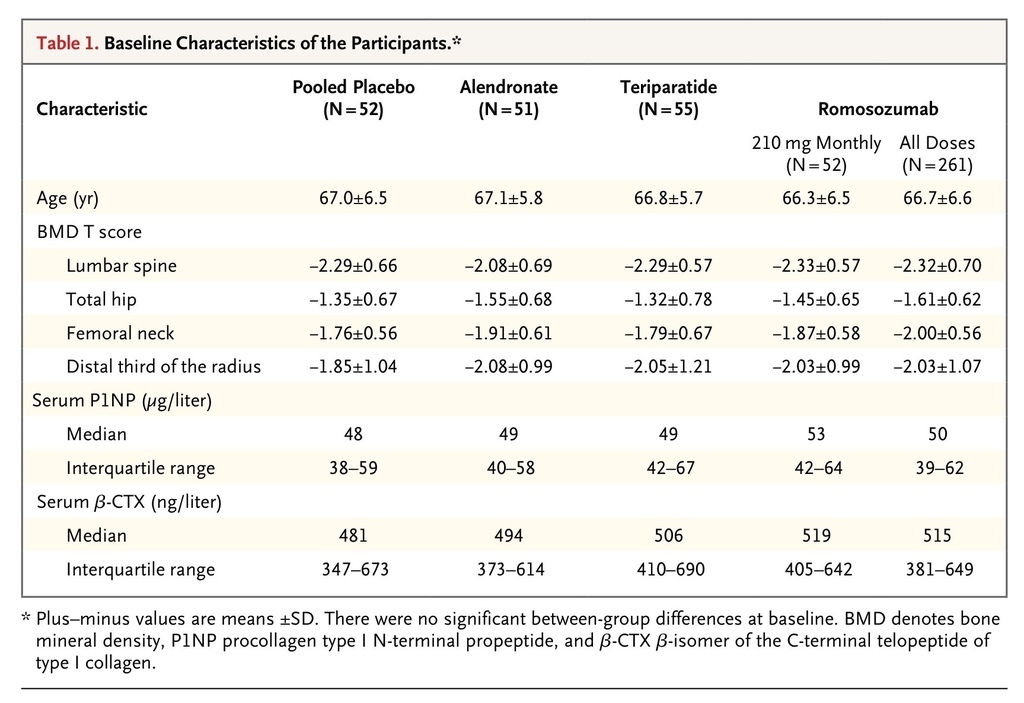
Baseline Characteristics of the Participants.
, and Table S1 in the Supplementary Appendix). The mean age of the participants was 67 years, 86% of the participants were white, and the mean T scores at the lumbar spine, total hip, and femoral neck were −2.29, −1.53, and −1.93, respectively.
Efficacy
Bone Mineral Density
At month 12, participants in the pooled romosozumab group, as compared with the pooled placebo group, had a significant increase in bone mineral density at the lumbar spine (primary end point; P<0.001), regardless of dose frequency (monthly or every 3 months) and dose level (140 mg or 210 mg) (Table S2 in the Supplementary Appendix). In addition, each of the five romosozumab groups, as compared with the pooled placebo group, had a significant increase in bone mineral density at the lumbar spine (Figure 2
FIGURE 2
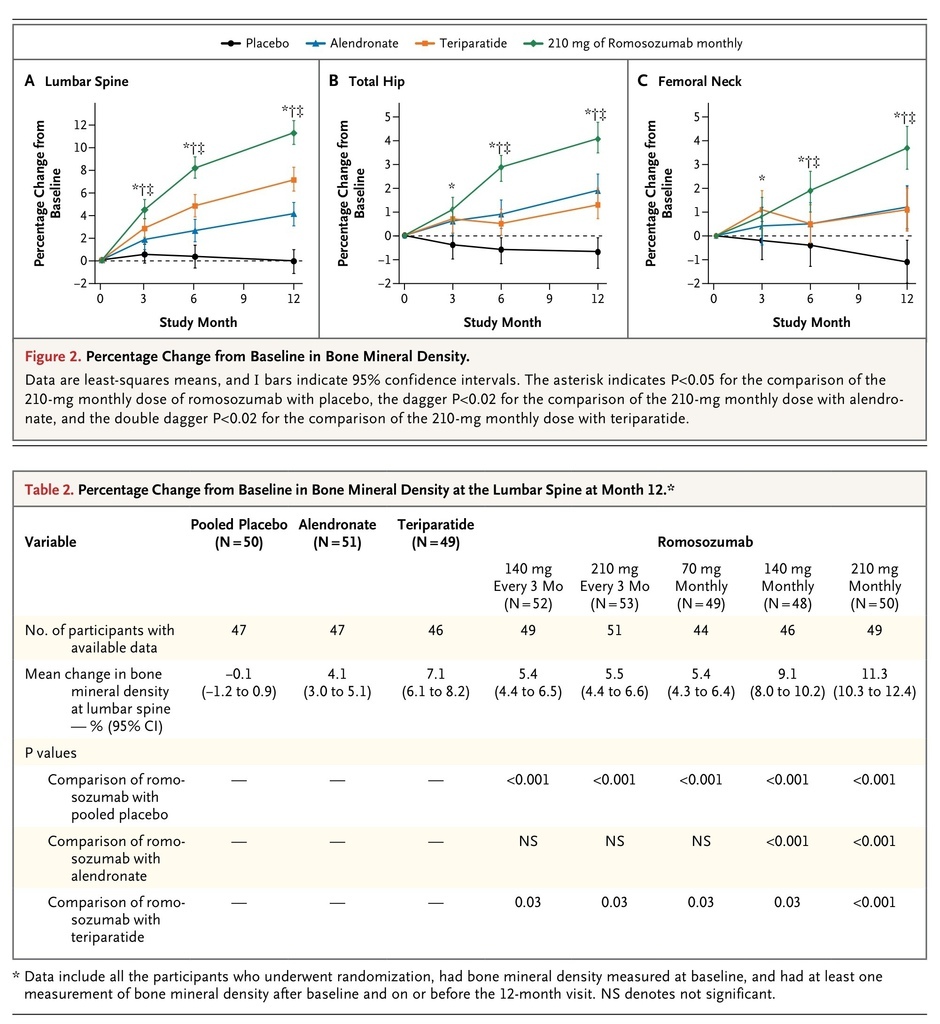
Percentage Change from Baseline in Bone Mineral Density.
and Table 2
Percentage Change from Baseline in Bone Mineral Density at the Lumbar Spine at Month 12.
, and Table S3 in the Supplementary Appendix), as well as at the total hip (Figure 2, and Table S4 in the Supplementary Appendix) and femoral neck (Figure 2, and Table S5 in the Supplementary Appendix) (P<0.001 for all comparisons).
The largest gains were observed with the 210-mg monthly dose of romosozumab, with mean increases from baseline to 12 months of 11.3% at the lumbar spine, 4.1% at the total hip, and 3.7% at the femoral neck. These increases were significantly greater than those observed in the alendronate and teriparatide groups (P<0.001 for all three comparisons) (Figure 2, and Tables S3, S4, and S5 in the Supplementary Appendix). No noteworthy differences in bone mineral density at the distal third of the radius were observed at 12 months between any of the romosozumab groups and the pooled placebo group, the alendronate group, or the teriparatide group (Table S6 in the Supplementary Appendix).
The bone mineral density at the lumbar spine and total hip was also significantly increased at month 6 in all the romosozumab groups as compared with the pooled placebo group (P≤0.006) (Figure 2, and Tables S3 and S4 in the Supplementary Appendix). The increases in bone mineral density at the femoral neck were significantly greater at month 6 in the groups that received romosozumab in doses of 140 mg monthly, 210 mg monthly, and 210 mg every 3 months than in the pooled placebo group (P<0.02) (Figure 2, and Table S5 in the Supplementary Appendix). Increases in bone mineral density at the lumbar spine, total hip, and femoral neck at month 6 were also significantly greater in the groups that received the two highest doses of romosozumab (140 mg monthly and 210 mg monthly) than in the groups that received alendronate or teriparatide (P≤0.01).
Markers of Bone Turnover
In all the romosozumab groups, increases in bone-formation markers were transitory. Increases were noted 1 week after the initial dose was administered and were greatest at month 1. The levels returned to baseline values or fell below baseline values between months 2 and 9, depending on the dose and the marker (Figure 3A
FIGURE 3
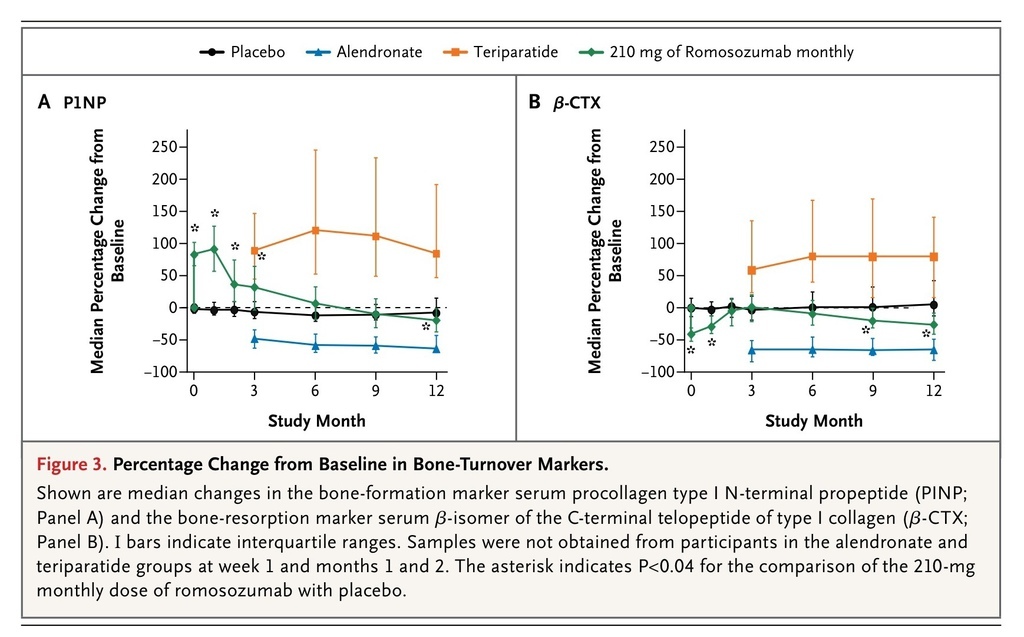
Percentage Change from Baseline in Bone-Turnover Markers.
, and Tables S7, S8, and S9 in the Supplementary Appendix).
In all the romosozumab groups, the level of the bone-resorption marker serum β-CTX initially decreased from baseline, with the largest median decrease apparent in the first week. In the groups that received monthly doses of romosozumab, the levels of serum β-CTX remained below baseline values at month 12 (Figure 3B, and Table S10 in the Supplementary Appendix).
Biochemical Analyses
Treatment with romosozumab was associated with a decrease from baseline in the serum calcium level (Table S11 in the Supplementary Appendix). The mean nadir was observed by month 1, at which time the change from baseline ranged from −0.03 to −0.07 mmol per liter (−0.12 to −0.28 mg per deciliter), representing a decrease of 1.30 to 2.68% from baseline, following a dose–response pattern. The serum calcium levels returned to baseline values at subsequent visits. Compensatory increases from baseline in the level of parathyroid hormone were observed in the romosozumab groups in a dose-dependent manner (Table S12 in the Supplementary Appendix). No adverse events of hypocalcemia were reported.
Adverse Events and Safety
The proportions of participants reporting adverse events and serious adverse events were similar in the pooled placebo group and the romosozumab groups (Table 3
TABLE 3
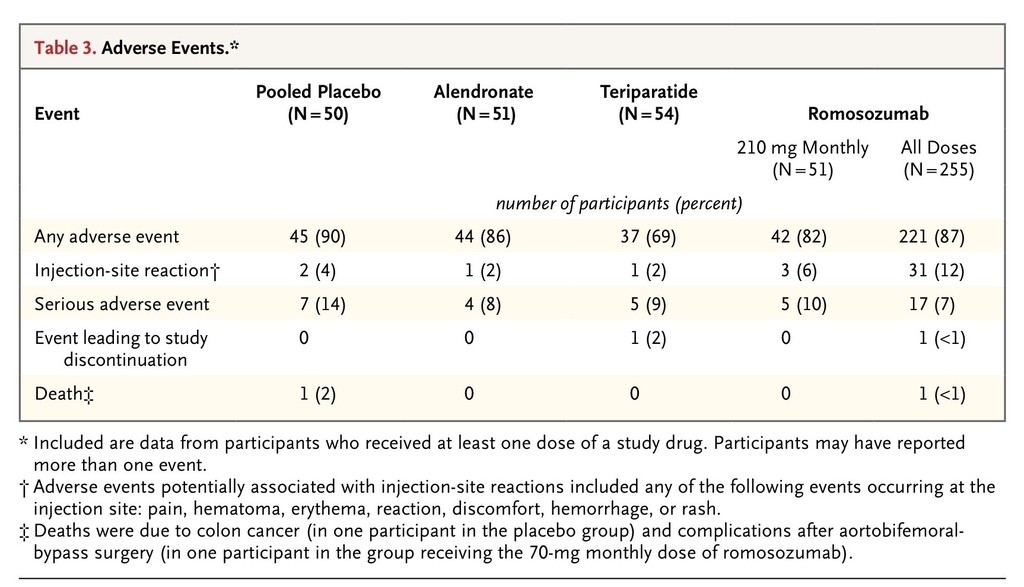
Adverse Events.
, and Table S13 in the Supplementary Appendix). No apparent relationship between dose and adverse events was observed. Injection-site reactions were observed more frequently with romosozumab than with placebo, but no dose–response relationship was observed. These reactions were generally mild, did not lead to discontinuation of the study drug or withdrawal from the study, and did not generally recur with continued administration of romosozumab.
The incidence of serious adverse events was 14% in the placebo group (7 of 50 participants), 8% in the alendronate group (4 of 51), 9% in the teriparatide group (5 of 54), 10% in the group that received the 210-mg monthly dose of romosozumab (5 of 51), and 7% across all romosozumab groups (17 of 255). Among the 5 participants with serious adverse events in the group that received the 210-mg monthly dose of romosozumab, the following events were reported (in 1 participant each): breast cancer, chronic obstructive pulmonary disease, noncardiac chest pain, wrist fracture (radius and ulna), and renal oncocytoma (benign).
No serious adverse event was reported by more than 1 participant in any group, and none of the serious adverse events were considered by the investigator to be treatment-related. One death due to colon cancer was reported in a participant who received placebo, and one death associated with a postoperative ileus (after aortobifemoral bypass) was reported in the group that received the 70-mg monthly dose of romosozumab.
No notable changes from baseline in vital signs, laboratory values, or electrocardiographic variables were noted in any of the participants. All the participants who received placebo, alendronate, or teriparatide tested negative for binding antibodies. Among participants who received romosozumab, binding antibodies were identified in 20% and antibodies with in vitro neutralizing activity in 3%. The development of antibodies had no discernible effect on adverse events, pharmacokinetics, or pharmacodynamics.
DISCUSSION
This study, which included 419 postmenopausal women with low bone mass, showed that inhibiting sclerostin with romosozumab, a humanized monoclonal antibody targeted to sclerostin, induced large increases in bone-formation markers, decreased a bone-resorption marker, and increased bone mineral density when the drug was administered by means of subcutaneous injection at 1-month or 3-month intervals. Although dose-dependency was not formally tested, the larger doses that were administered monthly (140 mg or 210 mg) appeared to produce greater changes than the other regimens. The response profiles of alendronate and teriparatide with respect to bone mineral density were as expected. The increase in bone mineral density at the lumbar spine and proximal femur was rapid and substantial with romosozumab by 3 months, and by 6 months the increase was greater with the 210-mg monthly dose of romosozumab than with either active comparator.
The effects of romosozumab on bone turnover reflect a rapid, marked, and transitory increase in bone formation and a moderate but more sustained decrease in bone resorption. These results confirm similar observations from the phase 1 clinical trial.13 The changes in bone-remodeling markers that were observed with romosozumab contrast with the effects of bisphosphonates and receptor activator of nuclear factor-κB ligand inhibitors, which reduce both bone-resorption and bone-formation markers. They also differ from the response to parathyroid hormone, with which the initial increase in bone-formation markers is followed by an increase in markers of bone resorption. The consequence of these divergent effects on bone formation and bone resorption with romosozumab is a strongly positive balance in bone turnover, accounting for the rapid and large increases in bone mineral density that we observed.
Circulating markers of bone formation increased rapidly with romosozumab but returned to baseline values despite continued administration, whereas a decrease in a circulating marker of bone resorption was maintained over the 12-month dosing period. The reason for the transitory nature of the effect on bone formation is unclear. Changes in counterregulatory signaling pathways would not be unexpected, given the complexity with which bone remodeling is controlled. No apparent relationship was observed between romosozumab antibodies and measures of efficacy. The implications of the transitory nature of the stimulation of bone formation on a dosing regimen for romosozumab in clinical practice will require more study. The specific mechanism responsible for the observed reduction in bone resorption with romosozumab is not understood.
Patients with lifelong homozygous or heterozygous genetic deficiency of sclerostin provide insight into the expected safety of inhibiting sclerostin signaling. Homozygous persons exhibit bony overgrowth and skeletal deformity, especially of the skull and face, which are observed during growth and which can result in symptoms related to compression of the VII and VIII cranial nerves.6,7 However, heterozygous carriers of the SOST mutation have increased bone density and modestly increased markers of bone formation but none of the sequelae of bony overgrowth.15,16 Therefore, potential complications due to bone overgrowth would not be expected with pharmacologic inhibition of sclerostin over a limited period of time in adults. Nevertheless, larger studies of romosozumab will be required in order to further evaluate the potential consequences of bone overgrowth.
The safety of drugs cannot be adequately evaluated in studies involving small treatment groups. In the current study, the overall incidence of adverse events was balanced between groups, with the exception of the increased frequency of injection-site reactions in the romosozumab groups as compared with the other groups. Numerical differences between the romosozumab groups and the control groups were noted with respect to the frequency of other reported adverse events, but some differences occurred in favor of romosozumab and others against romosozumab, and there was no relationship to the dose or to the timing of the event after administration of the drug.
In conclusion, romosozumab, administered subcutaneously at intervals of 1 month or 3 months over a period of 12 months, was associated with prompt, transitory increases in markers of bone formation; moderate, sustained decreases in markers of bone resorption; and rapid, large increases in bone mineral density in the spine and hip regions. The increases in bone mineral density were greater with romosozumab than with placebo, alendronate, or teriparatide. These results support further evaluation of romosozumab as a treatment for patients with osteoporosis.
如果你比較想要看機轉的圖:
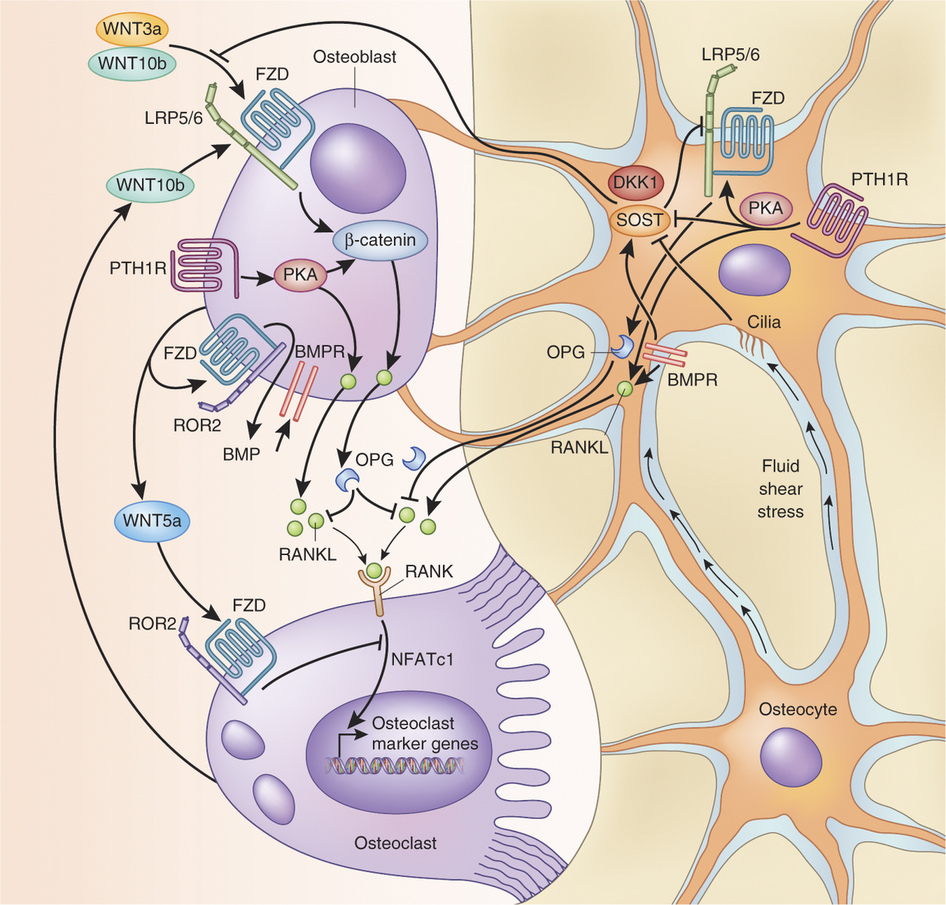
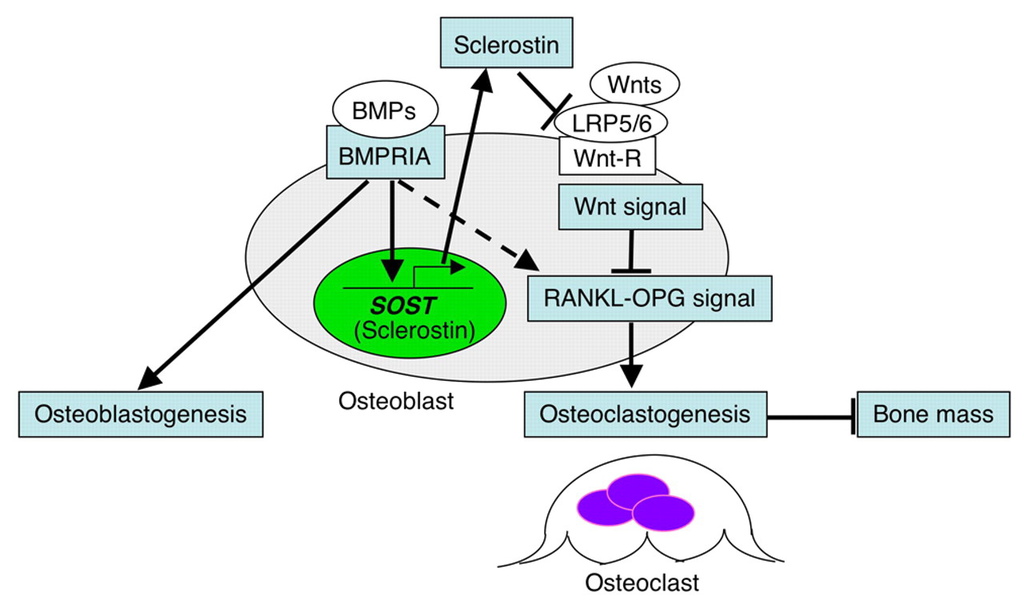


 留言列表
留言列表
 線上藥物查詢
線上藥物查詢 