

Chronic lymphocytic leukemia (CLL) is the most prevalent leukemia among adults. Standard treatments include combinations of purine analogues, alkylating agents, and monoclonal antibodies. In younger patients without major coexisting illnesses, these regimens can provide high response rates of durable length but have substantial toxic effects. As a result, these treatments often have unacceptable side effects in older patients and those with coexisting illnesses.1
Patients with relapsed CLL often have limited options because of the development of resistance to, or persisting toxic effects of, previous therapies. This is particularly true for elderly patients and those with coexisting illnesses.2 For these patients, the guidelines of the National Comprehensive Cancer Network recognize rituximab (Rituxan, Genentech/Biogen Idec) as a treatment option.3 Rituximab is commonly used in such patients, although it has not been approved as monotherapy. Rates of response to rituximab vary, and the duration of progression-free survival is generally short.4-7
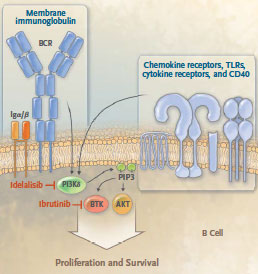
The B-cell–receptor signaling pathway plays a key role in the pathogenesis of CLL.8-11 Signaling through the B-cell receptor is mediated in part by the activation of the delta isoform of phosphatidylinositol 3-kinase (PI3Kδ). The delta isoform is one of four catalytic isoforms (p110 α, β, γ, and δ) that differ in their tissue expression, with PI3Kδ being highly expressed in lymphoid cells12 and the most critical isoform involved in the malignant phenotype in CLL.13 It activates the serine–threonine kinases AKT and mammalian target of rapamycin (mTOR) and exerts pleiotropic effects on cell metabolism, migration, proliferation, survival, and differentiation.14,15 Additional surface receptors that may play important roles in CLL pathophysiology (e.g., CXCR4,16 CD40,17 and CD49d18) also transduce their signals in part through PI3Kδ.
Idelalisib (formerly called GS-1101 and CAL-101) is a potent, oral, selective small-molecule inhibitor of PI3Kδ.15 In phase 1 studies, idelalisib (both as a single agent and in combination with other agents, including rituximab19,20) had clinically significant activity with an acceptable toxicity profile in patients with relapsed or refractory CLL. On the basis of these encouraging results, we conducted Study 116 — a phase 3, randomized, double-blind, placebo-controlled trial of combination therapy with idelalisib and rituximab in patients with relapsed CLL.
METHODS
Study Design and Conduct
The trial was designed by the sponsor, Gilead, with input from all the authors. Gilead implemented the study and provided data analysis. The trial was conducted, according to the principles of Good Clinical Practice, at 90 centers in the United States and Europe after approval by the relevant regulatory authorities and the institutional review board at each study site. All patients provided written informed consent. All the authors had full access to the study data and were involved in the interpretation of the data and in the preparation, revision, and final approval of the manuscript. All the authors made the decision to submit the manuscript for publication and vouch for the accuracy and completeness of the data and adherence to the study protocol (available with the full text of this article at NEJM.org).
Study Treatment
All patients were assigned to receive rituximab intravenously (at a dose of 375 mg per square meter of body-surface area), followed by 500 mg per square meter every 2 weeks for 4 doses and then every 4 weeks for 3 doses, for a total of 8 infusions. Patients were stratified according to the presence of 17p deletion (a deletion of the 17p13 chromosomal region in the gene encoding tumor-suppressor p53 [TP53]) or other TP53 mutations or the lack of somatic hypermutation in the gene encoding the immunoglobulin heavy-chain variable region (IGHV), all of which are associated with an inferior outcome. The patients were then randomly assigned to receive rituximab with either oral idelalisib (at a dose of 150 mg) (idelalisib group) or placebo (placebo group) twice a day.
Patients in the placebo group who had disease progression while enrolled in Study 116 could enroll in Study 117 to receive idelalisib. Patients in the idelalisib group who had disease progression could receive an increased dose of the drug (300 mg twice daily).
Eligibility
Eligible patients had CLL that had progressed within 24 months after their last treatment and were not able to receive cytotoxic agents for one or more of the following reasons: severe neutropenia or thrombocytopenia caused by cumulative myelotoxicity from previous therapies, an estimated creatinine clearance of less than 60 ml per minute, or a score on the Cumulative Illness Rating Scale (CIRS)21 of more than 6 for coexisting illnesses not related to CLL.2 (The CIRS score ranges from 0 to 56, with higher scores indicating an increased number or greater severity of coexisting illnesses.) Previous treatment must have included either a CD20 antibody–based regimen or at least two previous cytotoxic regimens.
Assessments
We scheduled clinic visits, which included laboratory testing, every 2 weeks for the first 12 weeks, then every 4 weeks for 12 weeks, and then every 6 weeks for 24 weeks, followed by visits every 12 weeks. The treatment response was assessed on the basis of serial computed tomography or magnetic resonance imaging of the neck, chest, abdomen, and pelvis (performed every 8 weeks for 6 months and every 12 weeks thereafter) and laboratory hematologic measurements.
We used the criteria of the International Workshop on Chronic Lymphocytic Leukemia (IWCLL)22 to determine rates of response and progression, including Richter's transformation (in which CLL is transformed into an aggressive lymphoma). These criteria were recently modified to exclude lymphocytosis as an isolated criterion for disease progression in patients treated with agents inhibiting the B-cell receptor.23,24 Independent review by a committee whose members were unaware of study-group assignments determined rates of response and disease progression for each patient and the dates of occurrence.
Centralized reference diagnostic procedures were performed at Ulm University (Ulm, Germany) or Cancer Genetics (Rutherford, NJ) with the use of fluorescence in situ hybridization for genomic aberrations, DNA sequencing for IGHV mutation status, and WAVE DNA fragment analysis and confirmatory Sanger sequencing for TP53 analyses, as described previously.25-27 Adverse events were graded with the use of the National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE), version 4.03.
End Points
The primary end point of the trial was progression-free survival. Secondary end points were rates of overall and complete response, lymph-node response, and overall survival.
Statistical Analysis
We calculated progression-free survival, which was defined as the interval from randomization to disease progression or death from any cause (whichever came first), using the Kaplan–Meier method and compared rates using a stratified log-rank test. We used a Cox model with adjustment for stratification to calculate hazard ratios. The rate of overall response was defined as the proportion of patients who had a complete or partial response on the basis of the IWCLL modified criteria.25 The lymph-node response rate was defined as the proportion of patients who had a decrease of 50% or more in lymphadenopathy. Overall survival was defined as the interval from randomization to death from any cause.
All efficacy analyses were based on the intention-to-treat principle unless otherwise stated. For binary-response end points, we used the Cochran–Mantel–Haenszel chi-square test, adjusted for stratification, to assess between-group differences. A sequential testing procedure was applied to adjust for the overall type I error rate — in other words, if the primary end point was significant, the secondary end points of rates of overall response, lymph-node response, and overall survival would be tested sequentially. The sample size provided a power of more than 85% to detect a 75% improvement in the median progression-free survival. Two interim analyses were prespecified after approximately 50% and 75% of the anticipated 119 events had occurred, at alpha levels of 0.001 and 0.005, respectively.
RESULTS
Patients
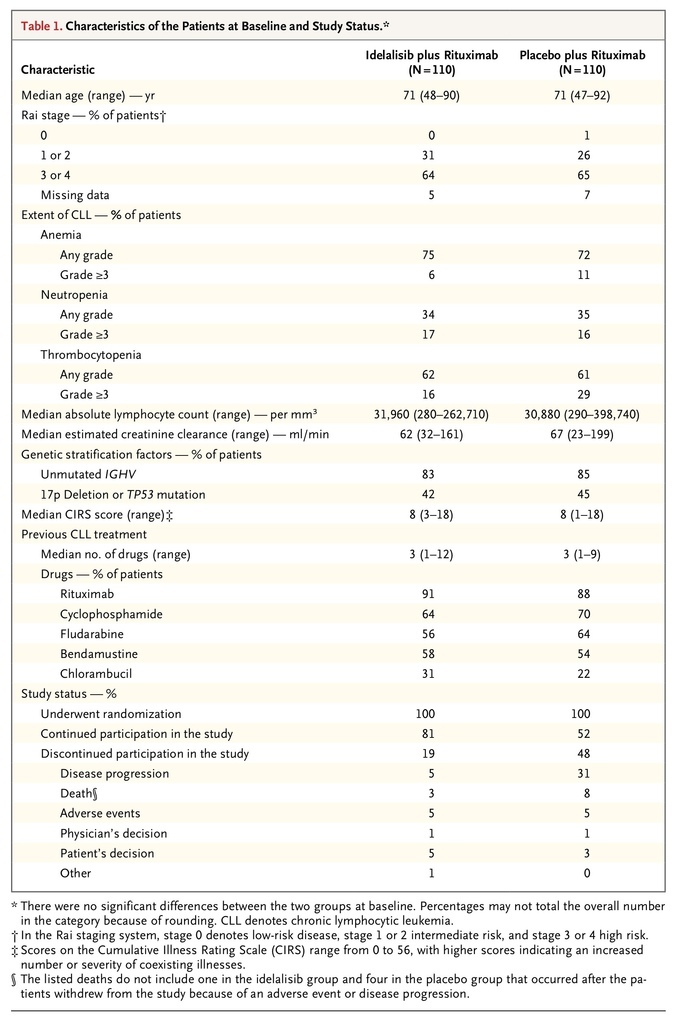
The 220 patients who were included in the study were enrolled between May 2012 and August 2013. The data cutoff for this analysis was August 30, 2013. A total of 78% of the patients were 65 years of age or older, 40% had at least moderate renal dysfunction (creatinine clearance, <60 ml per minute), 35% had poor bone marrow function (grade 3 or higher anemia, thrombocytopenia, or neutropenia), and 85% had a CIRS score of more than 6. The median CIRS score in each study group was 8 (Table 1
TABLE 1
Characteristics of the Patients at Baseline and Study Status.
).
Almost two thirds of the patients had advanced-stage disease, and the median time since the initial diagnosis of CLL was approximately 9 years. More than 80% of the patients had unmutated IGHV, and more than 40% had 17p deletion or TP53 mutations. Patients in the two study groups had received a median of three previous agents, including regimens containing rituximab, cyclophosphamide, fludarabine, and bendamustine.
A total of 110 patients were randomly assigned to each study group. All 110 patients in the idelalisib group and 107 in the placebo group actually received the assigned treatment (Table 1). Of the 3 patients in the placebo group who did not receive study treatment, 2 patients withdrew from the study because of an adverse event and 1 patient had not received study treatment before the data cutoff.
Receipt of Study Drug
At the time of this analysis, the median time that patients had received a study drug was 3.8 months (interquartile range, 1.9 to 8.6; simple range, 0.3 to 16+) in the idelalisib group and 2.9 months (interquartile range, 1.7 to 5.6; simple range, 0.1 to 14.6) in the placebo group. A total of 63 patients (39 in the idelalisib group and 24 in the placebo group) received a study drug for longer than 6 months. The duration of treatment was short for the two study groups owing to the recommendation of the data and safety monitoring board to stop the study. However, 81% of patients in the idelalisib group were still receiving the study drug at the time of study termination, as compared with only 52% in the placebo group. The most common reason for discontinuation of the study treatment was disease progression.
Efficacy
Progression-free Survival
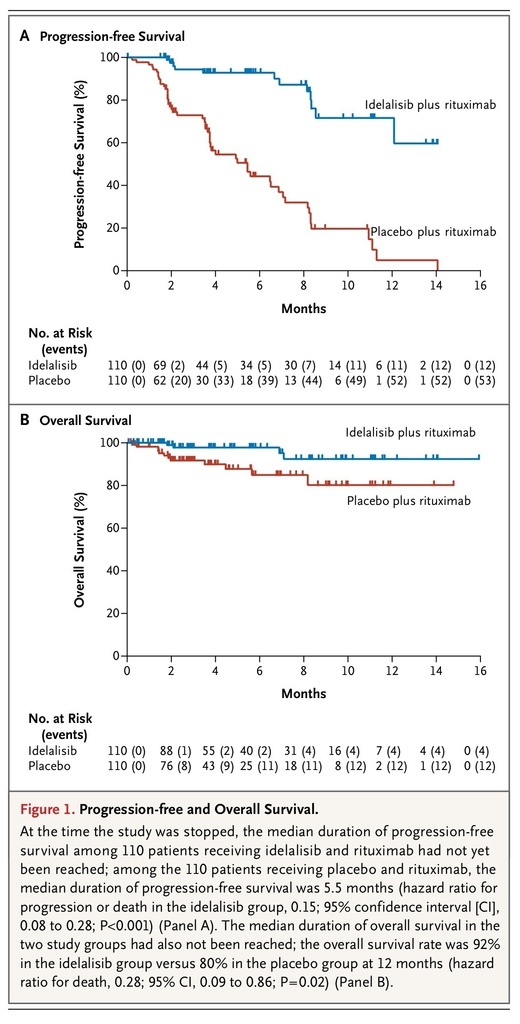
At 24 weeks, the rate of progression-free survival was 93% in the idelalisib group, as compared with 46% in the placebo group (adjusted hazard ratio for progression or death in the idelalisib group, 0.15; 95% confidence interval [CI], 0.08 to 0.28; unadjusted P<0.001, which crossed the prespecified stopping boundary for efficacy at the significance level of 0.001. Disease progression occurred in 12 patients in the idelalisib group and in 53 patients in the placebo group. The median duration of progression-free survival among patients in the idelalisib group was not reached, whereas the median progression-free survival was 5.5 months in the placebo group (Figure 1A
FIGURE 1
Progression-free and Overall Survival.
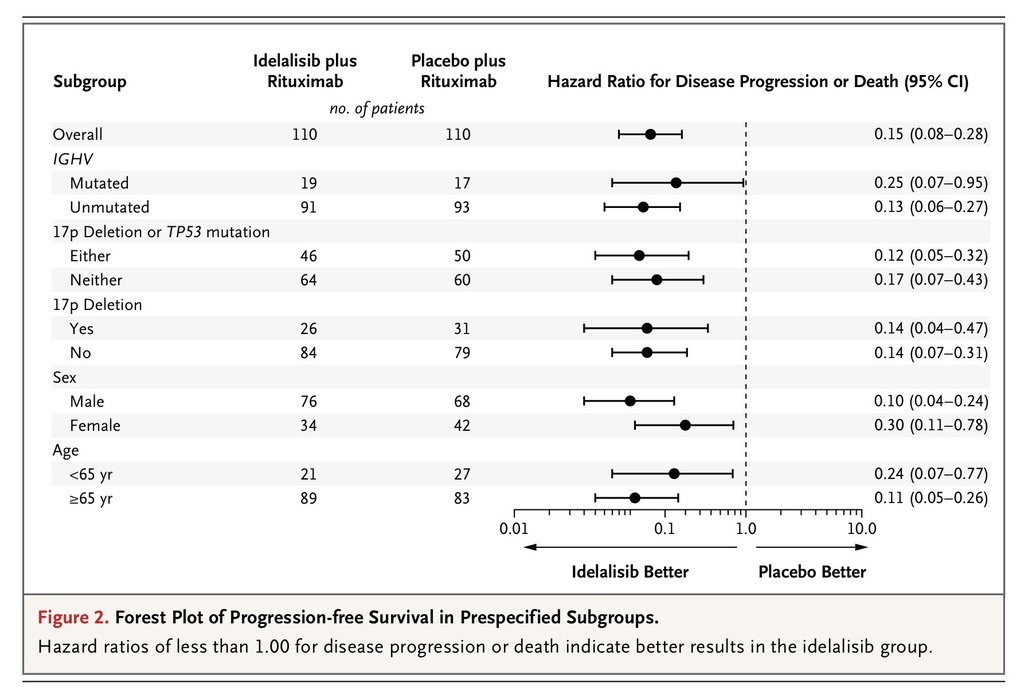
). The treatment effect of idelalisib and rituximab was similarly favorable in all prespecified subgroups, including those stratified according to the presence or absence of the 17p deletion or TP53 mutation and IGHV mutational status (Figure 2
FIGURE 2
Forest Plot of Progression-free Survival in Prespecified Subgroups.
).
Overall Survival
The rate of overall survival in the idelalisib group was superior to that in the placebo group (92% vs. 80% at 12 months), with an adjusted hazard ratio for death of 0.28 (95% CI, 0.09 to 0.86; P=0.02). The median duration of overall survival in the two groups had not yet been reached at the time of analysis (Figure 1B). Sixteen patients died while participating in the study: 4 patients (4%) in the idelalisib group and 12 patients (11%) in the placebo group.
Overall Response
The rate of overall response was evaluated for the 176 patients (88 patients in each study group) who had undergone at least one post-baseline assessment or had discontinued the study before the first assessment at the time of this analysis. The overall response rate was 81% (95% CI, 71 to 88) in the idelalisib group, as compared with 13% (95% CI, 6 to 21) in the placebo group (odds ratio, 29.92; P<0.001). All responses were partial responses.
Lymph-Node Response
A total of 169 patients underwent at least one post-baseline imaging assessment of the lymph-node response to treatment. On the basis of a review of imaging results by the independent review committee, the proportion of patients with a reduction of 50% or more in lymphadenopathy was significantly higher in the idelalisib group than in the placebo group (93%; 95% CI, 85 to 97; vs. 4%; 95% CI, 1 to 10), for an odds ratio of 264 (P<0.001) (Figure 3A
FIGURE 3
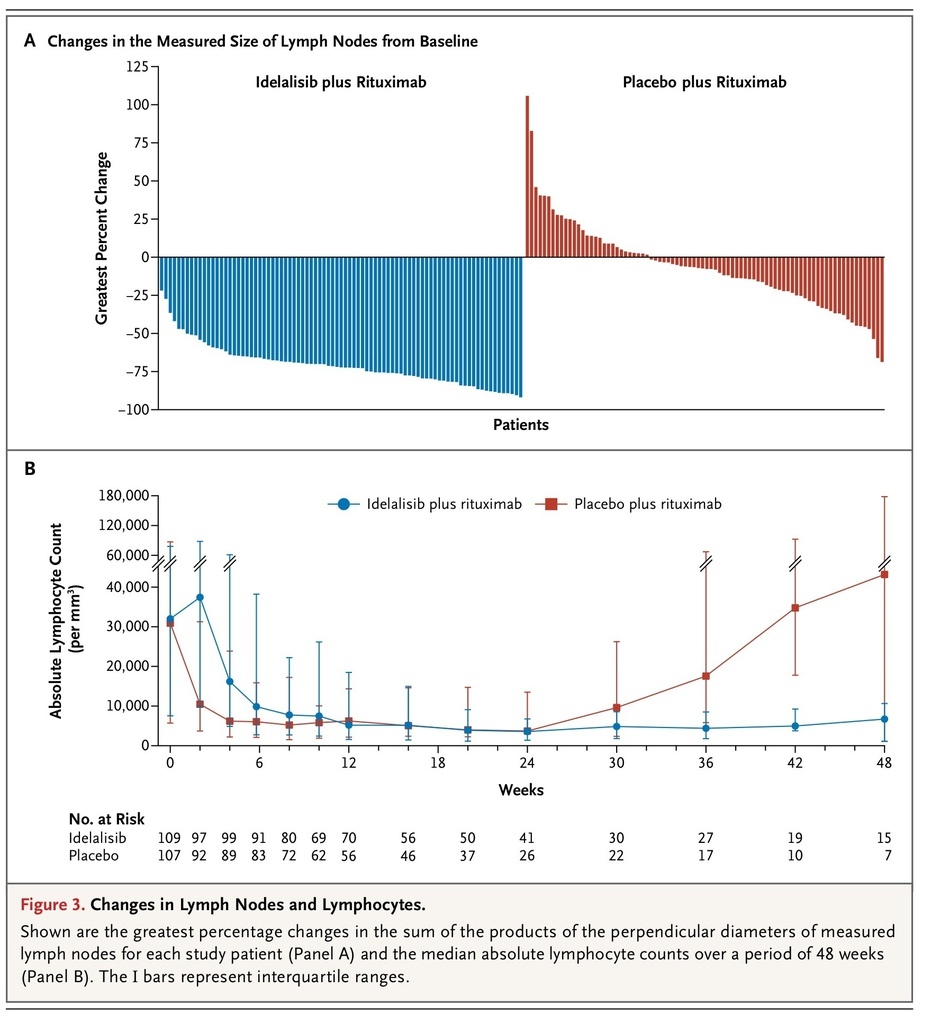
Changes in Lymph Nodes and Lymphocytes.
).
Idelalisib-Associated Lymphocytosis
Idelalisib, like other novel agents targeting the B-cell–receptor signaling pathway, has been shown to cause lymphocytosis when it is administered as a single agent.19 The addition of rituximab to idelalisib blunted and shortened the duration of the lymphocytosis, which confirmed the findings of a previous phase 1 study.20 The rate of lymphocytosis peaked at week 2 and resolved by week 12 in the idelalisib group (Figure 3B). In contrast, there was a sustained increase in the absolute lymphocyte count in the placebo group starting at week 24, which coincided with the completion of rituximab therapy. As per the modified IWCLL guidelines, this rise in the lymphocyte count was not considered to be disease progression.
Adverse Events
Adverse events, serious adverse events, and laboratory abnormalities that occurred during treatment are listed in Table 2
TABLE 2
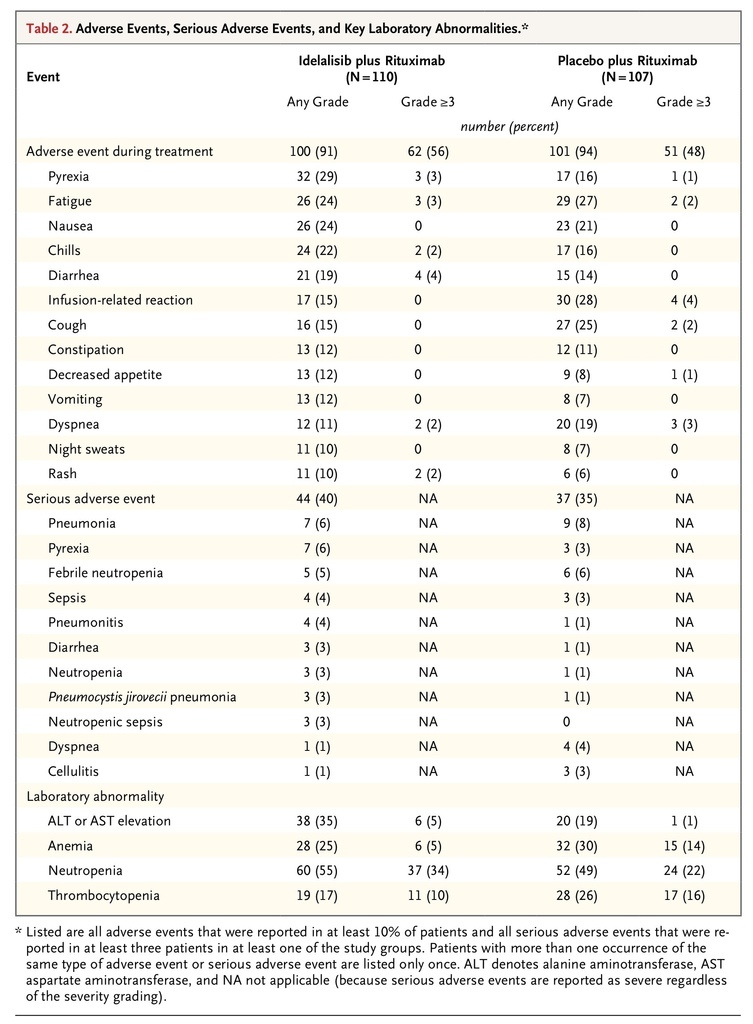
Adverse Events, Serious Adverse Events, and Key Laboratory Abnormalities.
. Most of the adverse events were consistent with those expected for a population with relapsed CLL that had received extensive prior therapy.
More than 90% of the patients had at least one adverse event. In the idelalisib group, the five most common adverse events were pyrexia, fatigue, nausea, chills, and diarrhea. In the placebo group, the adverse events were similar to those in the idelalisib group, with the most common being infusion-related reactions, fatigue, cough, nausea, and dyspnea. Most adverse events across the two study groups were grade 2 or lower. In the idelalisib group, grade 3 or higher diarrhea was reported in four patients and grade 3 or higher rash was reported in two patients, with no grade 3 or higher diarrhea or rash reported in the placebo group.
Common laboratory abnormalities included anemia, neutropenia, and thrombocytopenia. Hepatic aminotransferase elevations occurred more frequently in patients receiving idelalisib and rituximab than in those receiving placebo and rituximab. Grade 3 or higher elevations occurred in six patients (5%) in the idelalisib group, with onset at 8 to 16 weeks. The study drug was withheld and successfully reinitiated in four of these six patients. No patients discontinued the study drug because of aminotransferase elevations.
At least one serious adverse event occurred in 44 patients (40%) in the idelalisib group and in 37 patients (35%) in the placebo group. The most frequent serious adverse events in the two groups were pneumonia, pyrexia, and febrile neutropenia.
Adverse events leading to study-drug discontinuation were reported in 9 patients (8%) in the idelalisib group and 11 patients (10%) in the placebo group. In the idelalisib group, gastrointestinal and skin disorders contributed to 6 discontinuations. In the placebo group, infections and respiratory disorders contributed to 8 discontinuations.
DISCUSSION
In our study involving patients with relapsed CLL who were not able to undergo standard cytotoxic chemotherapy, treatment with idelalisib and rituximab was associated with significant improvement in the rate of progression-free survival, as compared with placebo and rituximab, a finding that warranted termination of the study after the first prespecified interim analysis. We chose rituximab monotherapy as the comparator for this study on the basis of National Comprehensive Cancer Network guidelines and clinical-use data showing that the drug is a commonly prescribed treatment in the United States for this population and is increasingly being prescribed in Europe (see the Supplementary Appendix, available at NEJM.org). The patients were defined as less able to receive cytotoxic chemotherapy on the basis of severe myelosuppression from previous chemotherapy, reduced kidney function, or a CIRS score of more than 6 for non–CLL-related coexisting illnesses. We used a schedule for the administration of rituximab that was approved in combination with fludarabine and cyclophosphamide; two additional doses were added to achieve dose intensification.
Improvement in progression-free survival was observed not only in the overall study population but also in all subgroups examined, including patients with poor prognostic features, such as 17p deletion or TP53 mutations and unmutated IGHV. The rate of progression-free survival in the placebo group was similar to that reported for rituximab monotherapy in other trials involving patients who were not selected on the basis of medical fitness for chemotherapy. Furthermore, the median duration of progression-free survival among patients receiving placebo and rituximab was similar to the range of 5.7 to 5.9 months reported among patients receiving monotherapy with ofatumumab, another anti-CD20 antibody approved as monotherapy for disease that is resistant to fludarabine and alemtuzumab.28 In addition, patients receiving idelalisib and rituximab had significant improvement in overall survival, as compared with those receiving placebo and rituximab. This improvement in survival is further evidence of the clinical importance of the progression-free survival benefit shown in this trial. Significant differences favoring the idelalisib group were also observed in rates of overall response and lymph-node response.
There was no overall increase in the rate of adverse events with the addition of idelalisib to rituximab, as compared with placebo and rituximab. Grade 3 or 4 adverse events were common in the two study groups, which was an expected finding, given the large number of previous treatments, clinically significant coexisting illnesses, and prolonged time since the initial diagnosis of CLL in these patients. In earlier studies, idelalisib was associated with elevated aminotransferase elevations, rash, and severe diarrhea.19 These adverse events were reported in the idelalisib group in our study as well, albeit at a low frequency, and severe events were generally managed with study-drug interruption and symptom treatment. Since the study was stopped early for efficacy, severe diarrhea, which is typically a late-onset event, may yet occur in some patients. A surprising finding was a reduction in rates of infusion-related toxicity from rituximab in the idelalisib group.
In conclusion, the addition of idelalisib to rituximab in a population of frail, difficult-to-treat patients, including those with adverse genetic features such as 17p deletion or TP53 mutations or unmutated IGHV, was superior to rituximab monotherapy, which is commonly used in such patients. Although the follow-up in this study was short, combination therapy with idelalisib had an acceptable safety profile. Further follow-up is needed to assess whether idelalisib is safe for long-term use. Patients with CLL who are less able to undergo standard chemotherapy are frequently excluded from clinical trials because of the presence of coexisting illnesses, yet such patients are often seen in clinical practice. On the basis of response rates and progression-free survival results, the combination of idelalisib and rituximab may be a treatment option for these patients. Idelalisib is part of a growing list of agents with activity in CLL, including ibrutinib (targeting Bruton's tyrosine kinase)29 and ABT-199 (targeting B-cell lymphoma 2 protein [BCL2]).30 Additional studies will be necessary to define the most effective use of these new agents.
Presented in part at the annual meeting of the American Society of Hematology, New Orleans, December 7–10, 2013.
Supported by Gilead.
Disclosure forms provided by the authors are available with the full text of this article at NEJM.org.
Drs. Furman, Sharman, and Coutre contributed equally to this article.
This article was published on January 22, 2014, at NEJM.org.
We thank the patients who participated in this study; the investigators and coordinators at the clinical sites; the employees of Gilead who contributed to the design, implementation, and data analysis; Eugen Tausch, M.D., for the genetic analysis; and Tim DiChiara, Ph.D., for assistance in the preparation of the manuscript.


 留言列表
留言列表
 線上藥物查詢
線上藥物查詢 