

70-07 節錄
Dalbavancin, oritavancin and telavancin are semisynthetic lipoglycopep- tides that demonstrate promise for the treatment of patients with infections caused by multi-drug-resistant Gram-positive pathogens. Each of these agents contains a heptapeptide core, common to all glycopeptides, which enables them to inhibit transglycosylation and transpeptidation (cell wall synthesis). Modifications to the heptapeptide core result in different in vitro activities for the three semisynthetic lipoglycopeptides. All three lipoglyco- peptides contain lipophilic side chains, which prolong their half-life, help to anchor the agents to the cell membrane and increase their activity against Gram-positive cocci. In addition to inhibiting cell wall synthesis, telavancin and oritavancin are also able to disrupt bacterial membrane integrity and increase membrane permeability; oritavancin also inhibits RNA synthesis. Enterococci exhibiting the VanA phenotype (resistance to both vancomycin and teicoplanin) are resistant to both dalbavancin and telavancin, while oritavancin retains activity. Dalbavancin, oritavancin and telavancin exhibit activity against VanB vancomycin-resistant enterococci. All three lipoglyco- peptides demonstrate potent in vitro activity against Staphylococcus aureus and Staphylococcus epidermidis regardless of their susceptibility to meticillin, as well as Streptococcus spp. Both dalbavancin and telavancin are active against vancomycin-intermediate S. aureus (VISA), but display poor activity versus vancomycin-resistant S. aureus (VRSA). Oritavancin is active against both VISA and VRSA. Telavancin displays greater activity against Clos- tridium spp. than dalbavancin, oritavancin or vancomycin.
The half-life of dalbavancin ranges from 147 to 258 hours, which allows for once-weekly dosing, the half-life of oritavancin of 393 hours may allow for one dose per treatment course, while telavancin requires daily adminis- tration. Dalbavancin and telavancin exhibit concentration-dependent ac- tivity and AUC/MIC (area under the concentration-time curve to minimum inhibitory concentration ratio) is the pharmacodynamic parameter that best describes their activities. Oritavancin’s activity is also considered concentra- tion-dependent in vitro, while in vivo its activity has been described by both concentration and time-dependent models; however, AUC/MIC is the phar- macodynamic parameter that best describes its activity.
Clinical trials involving patients with complicated skin and skin structure infections (cSSSIs) have demonstrated that all three agents are as efficacious as comparators. The most common adverse effects reported with dalbavancin use included nausea, diarrhoea and constipation, while injection site reac- tions, fever and diarrhoea were commonly observed with oritavancin ther- apy. Patients administered telavancin frequently reported nausea, taste disturbance and insomnia. To date, no drug-drug interactions have been identified for dalbavancin, oritavancin or telavancin. All three of these agents are promising alternatives for the treatment of cSSSIs in cases where more economical options such as vancomycin have been ineffective, in cases of reduced vancomycin susceptibility or resistance, or where vancomycin use has been associated with adverse events.
Fig. 1. Structure activity relationships of the glycopeptides. DAL = dalbavancin; ORI = oritavancin; TEL = telavancin; VAN = vancomycin.
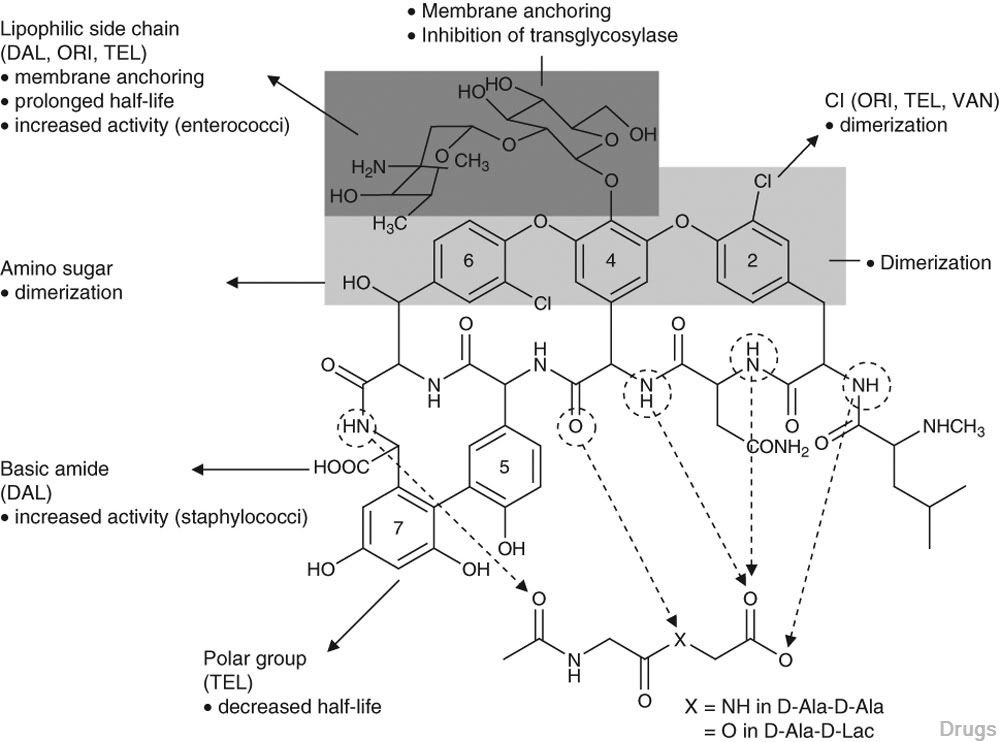
Fig. 2. Chemical structures of dalbavancin, oritavancin, telavancin and vancomycin.
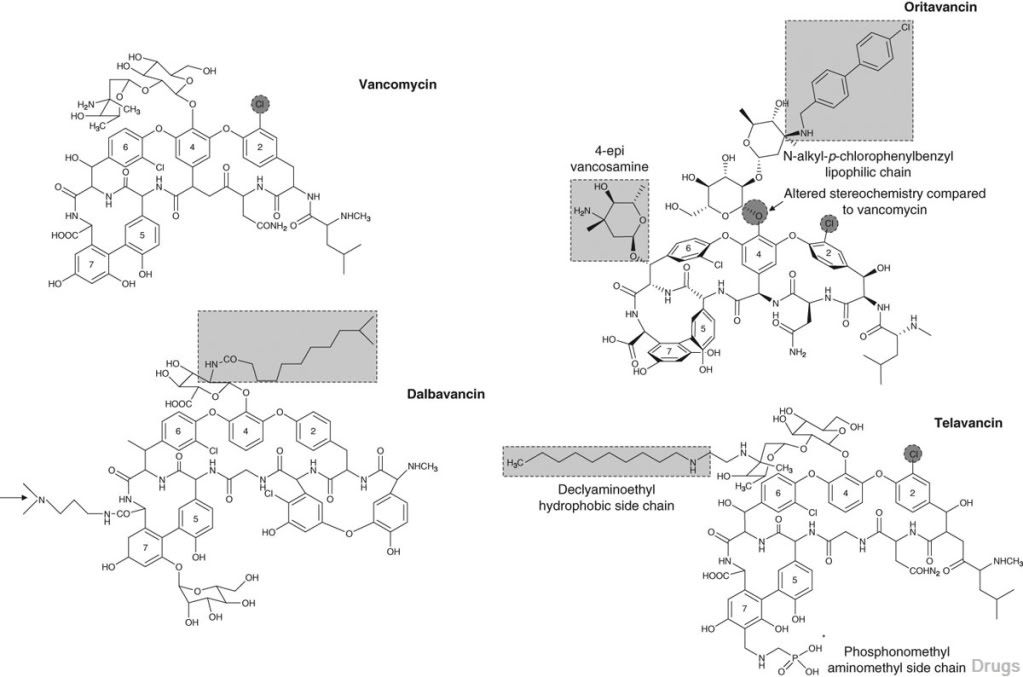
Table I. In vitro activity of dalbavancin, oritavancin, telavancin and vancomycin against Gram-positive organisms
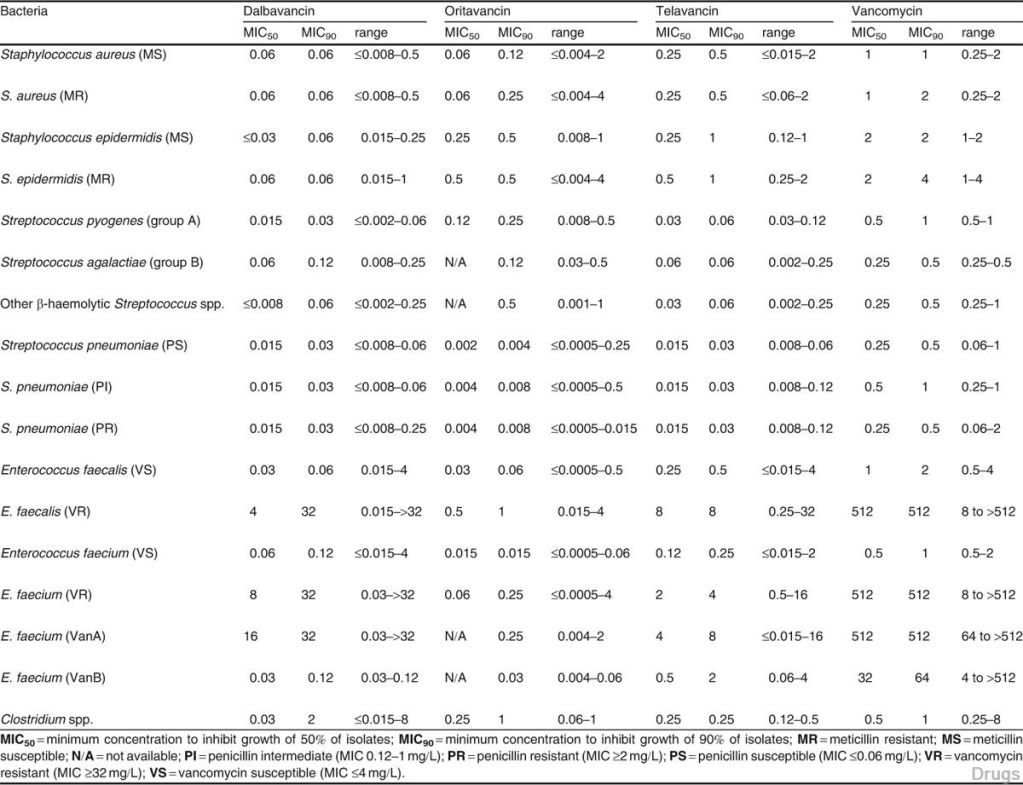
Table II. Pharmacokinetic parameters for glycopeptides at usual human doses (data obtained from healthy volunteers)
Table III. Summary of selected clinical trials of dalbavancin, oritavancin and telavancin invarious infections

Mechanism of Action
The mechanism of action common to all members of the glycopeptide class is the inhibi- tion of bacterial cell wall synthesis.Glycopeptides bind to the terminal D-Ala-D-Ala in growing peptidoglycan chains, and through this binding hinder polymerization and cross-linking ultimately resulting in the destabilization of the cell wall, with bacterial cell death occurring pre- sumably due to osmotic insult. The chemical structures of the different lipoglycopeptide agents impart additional mechanisms of action to cer- tain members of the group, including oritavancin and telavancin (figure 1). A feature common to the newer lipoglycopeptides is the presence of lipid side chains, which anchor the molecule to the cell membrane and thereby concentrate the drug at its site of action, dramatically increasing their potency relative to their parent glycopep- tide. In addition, it has been proposed that the lipid side chains may act as detergent- like molecules and cause partial destabilization of the cell membrane and loss of membrane potential.
Dalbavancin
Dalbavancin inhibits cell wall synthesis in Gram-positive bacteria through the formation of a stable complex between its heptapeptide back- bone and the D-Ala-D-Ala portion of cell wall precursors. This complex prevents the trans- glycosylase and transpeptidase enzymes from polymerizing and cross-linking the peptidogly- can, and as a result the peptidoglycan chain growth is arrested. In addition, it appears that the lipophilic side chain of dalbavancin enhances the antibacterial’s binding affinity for its D-Ala- D-Ala target site through the formation of dimers and membrane anchoring, leading to a superior potency than its parent glycopeptide.Dimer- ization aids the process by maintaining the bind- ing pocket in the optimal position for cooperative binding, while membrane anchoring helps by focusing dalbavancin closer to its target. The role of the lipid side chain in destabilization of cell membranes has not been demonstrated for dalbavancin.
Oritavancin
Oritavancin inhibits cell wall synthesis by complexing with the terminal D-Ala-D-Ala of a nascent peptidoglycan chain and also to the pentaglycine bridge, thus inhibiting transglyco- sylation and transpeptidation. Unlike other glycopeptides, oritavancin is able to bind to depsipeptides including D-Ala-D-Lac, which fa- cilitates its inhibition of cell wall synthesis even in organisms exhibiting VanA-type resistance. Oritavancin forms homodimers prior to binding to D-Ala-D-Ala or D-Ala-D-Lac, which increases its binding affinity for the target site.The p-chloro-phenylbenzyl side chain of oritavancin interacts with the cell membrane, exerting two beneficial effects. This binding acts to main- tain the antibacterial in a prime position for peptidoglycan interactions and it also imparts oritavancin with the ability to disrupt the bac- terial membrane potential and thus increase membrane permeability.[22,23] Oritavancin has been shown to dissipate membrane potential in both stationary and exponential phase growing bacteria, which is rare and may carry clinical implications in terms of its activity against slowly growing organisms and biofilms. The dual mechanism of action could also theoretically increase effectiveness and reduce the risk of resist- ance selection. In addition to the aforemen- tioned mechanisms, it has also been hypothesized that oritavancin inhibits RNA synthesis.
Telavancin
Telavancin exhibits a dual mechanism of ac- tion against Gram-positive bacteria. As with other glycopeptides, telavancin binds to terminal D-Ala-D-Ala residues and inhibits cell wall synthesis. Due to its ability to bind to bacterial cell membranes, telavancin is ten times more active than vancomycin at inhibiting both the transglycosylation and the synthesis of pep- tidoglycan.Like oritavancin, telavancin also alters cell membrane permeability and dissipates membrane potential. This second mechanism of action may promote or be the lone reason for its rapid bactericidal activity.
Mechanisms of Resistance
Numerous mechanisms of resistance have been elucidated for glycopeptides, which account for resistance or reduced susceptibility to vanco- mycin.[26,27] A number of glycopeptide-resistant phenotypes have been found, and these include VanA, VanB, VanC, VanD, VanE and VanG types.[27] VanA is the most common type and is responsible for high-level resistance to both van- comycin and teicoplanin.[27] These mechanisms of resistance arise via alteration of peptidoglycan precursors such that pentapeptides normally ter- minating in D-Ala-D-Ala are revised to terminate in D-Ala-D-Lac or D-alanine-D-serine.[27] This change results in the loss of one of five hydrogen bonds between the pentapeptide core of the gly- copeptide and its target, and thus reduces the affinity of the antibacterial for its target.[27] In the case of an alteration to D-Ala-D-Lac, the affinity of the glycopeptide for the target may be reduced up to 1000-fold.[2,27] Expression of the vanA and vanB operons is inducible and expression is dri- ven by glycopeptides. Vancomycin activates both the vanA and vanB operons, while teicoplanin, oritavancin, dalbavancin and telavancin are only activators of the vanA operon (table I).[27] As a result, teicoplanin, oritavancin, dalbavancin and telavancin retain activity against strains that express VanB.[27]
VanA-type, and to a lesser extent VanB-type, acquired resistance is common among en- terococci due to their ability to easily acquire and exchange genetic material.[24] Interspecies gene transfer is also possible and clinically important as it has been reported that the Michigan and Pennsylvania clinical VRSA isolates resulted from an in vivo transfer of the vanA transposon from Enterococcus faecalis (it can also occur with Enterococcus faecium) to MRSA.[24] In addition, VanA and VanB have rarely been identified in other Gram-positive bacteria, including Coryne- bacterium spp., Arcanobacterium haemolyti- cum, Lactococcus spp., Streptococcus bovis and anaerobes.[2] VanC is typically associated with the intrinsically vancomycin-resistant Enterococcus casseliflavus and Enterococcus gallinarum. VanC is constitutively expressed and confers low-level re- sistance to vancomycin but not teicoplanin. The relatively higher potency of the newer lipoglycoli- popeptides may well overcome this low-level resis- tance, the clinical relevance of which is not known. VanD, E, G and L are rare genotypes of glyco- peptide resistance infrequently encountered in clinical practice. The activity of the new lipoglyco- lipopeptides against these strains is not well known.
Resistance to vancomycin in the Mu50 strain of S. aureus may be attributed to a thickening of the cell wall, accelerated peptidoglycan synthesis and reduced peptidoglycan cross-linking.[49] Changes in teichoic acids of the cell wall may also be important in this mechanism of resistance.[2] Reduced susceptibility or resistance to vanco- mycin in VISA strains results from an excess of D-Ala-D-Ala residues, which bind the anti- bacterial and thus prevent it from reaching its target.[49] Intrinsic resistance to glycopeptides in uncommon pathogens including Pediococcus, Leuconostoc and Lactobacillus spp. is mediated by a D-alanine:D-lactate ligase that constitutively produces peptidoglycan precursors that end in D-Ala-D-Lac.
Dalbavancin-Resistance Selection
Resistance to dalbavancin occurs as a result of the aforementioned change in peptidoglycan precursors from vanA-expressed D-Ala-D-Ala to D-Ala-D-Lac.[3] The presence of the vanB operon does not confer resistance to dalbavancin, since the latter is not an inducer of the vanB operon. There has been no single-step, high-level dalba- vancin resistance (frequency <10-10) detected when Staphylococcus haemolyticus and S. aureus were plated on agar containing 10 mg/L of dal- bavancin.[2,14] The lack of single-step dalbavancin resistance was also demonstrated in MRSA and VISA isolates when incubated at 0.5, 1, 2, 4 and 8 times the MIC of dalbavancin.[14] Following 24 serial passages of S. aureus and S. haemolyticus at sub-MIC concentrations of dalbavancin, the dalbavancin MICs increased 2-fold for S. aureus and 4-fold for S. haemolyticus; however, the rea- son for this increase is not yet known.[14] Upon examination of the subcultures of colonies from different passage steps, it was shown that there was little variability in the dalbavancin MIC dis- tribution, whereas the vancomycin MICs dis- played heterogeneity.[2] These preliminary data suggest that the selection of dalbavancin resis- tance among staphylococci is slower than that with vancomycin.
Oritavancin-Resistance Selection
Although clinically isolated VanA, VanB and VanC enterococci are susceptible to oritavancin, it has been demonstrated in vitro that moderate- level resistance to oritavancin can occur in en- terococci isolates exhibiting the VanA or VanB phenotypes.[26] However, resistance in isolates harbouring the vanB operon were only observed when the operon was altered such that it became inducible by teicoplanin or constitutively ex- pressed.[26] Isolates producing peptidoglycan precursors terminating in D-Lac may exhibit re- sistance to oritavancin if all precursors terminat- ing in D-Ala are eliminated.[26] This has been achieved in vitro by increasing resistance gene expression or reducing D-Ala-D-Ala produc- tion.[26] Such changes resulted in a moderate-level resistance to oritavancin (MIC £16mg/L).[26,27] The expression of the vanZ gene of the vanA gene cluster also resulted in resistance to oritavancin, with an increase to an MIC of 8 mg/L through an unknown mechanism.[26] Thus far it has not been possible to select for high-level oritavancin resis- tance in the laboratory and there have been no cases of resistance to this lipoglycopeptide drug among clinically isolated pathogens.
Telavancin-Resistance Selection
In vitro, telavancin has been reported to have a low incidence of spontaneous resistance devel- opment among enterococci and staphylococci. It has been demonstrated that the frequency of spontaneous resistance to telavancin was <4.0 · 10-11 to <2.9 · 10-10 at 2 · MIC for MRSA and VISA, which was lower than the spontaneous mutation frequencies obtained with comparators.


 留言列表
留言列表
 線上藥物查詢
線上藥物查詢 