

John V. Fahy, M.D., and Burton F. Dickey, M.D.
The lungs are remarkably resistant to environmental injury, despite continuous exposure to pathogens, particles, and toxic chemicals in inhaled air. Their resistance depends on a highly effective defense provided by airway mucus,1-7 an extracellular gel in which water and mucins (heavily glycosylated proteins) are the most important components. Airway mucus traps inhaled toxins and transports them out of the lungs by means of ciliary beating and cough
Paradoxically, although a deficient mucous barrier leaves the lungs vulnerable to injury, excessive mucus or impaired clearance contributes to the pathogenesis of all the common airway diseases.1-4 This review examines the normal formation and clearance of airway mucus, the formation of pathologic mucus, the failure of mucus clearance that results in symptoms and abnormal lung function, and the therapy of mucus dysfunction.
STRUCTURE AND FUNCTION OF THE NORMAL AIRWAY
Epithelial surfaces in contact with the outside environment are protected by mechanical barriers (e.g., keratinized skin) and chemical barriers (e.g., gastric acid). Mucosal surfaces are wet epithelia that have a mucous barrier as part of their protective mechanism.1-7Mucus layers vary widely in composition and structure; for example, they are thick and adherent to the epithelium in the gut, but thin and mobile in the airway.
Surface Epithelial Cells
The surface epithelium of intrapulmonary airways is composed of two principal cell types — ciliated and secretory

These cells are present in similar numbers and form a mosaic. Secretory cells have been further divided into subtypes based on their microscopical appearance (e.g., Clara, goblet, and serous cells). However, studies indicate great structural, molecular, and functional plasticity in secretory cells.10-14 Therefore, it is simplest to refer to them generically as “secretory cells.” Besides mucins, secretory cells release a variety of antimicrobial molecules (e.g., defensins, lysozyme, and IgA), immunomodulatory molecules (e.g., secretoglobins and cytokines), and protective molecules (e.g., trefoil proteins and heregulin) constitutively and inducibly; these can become incorporated into mucus.15,16
Submucosal Glands
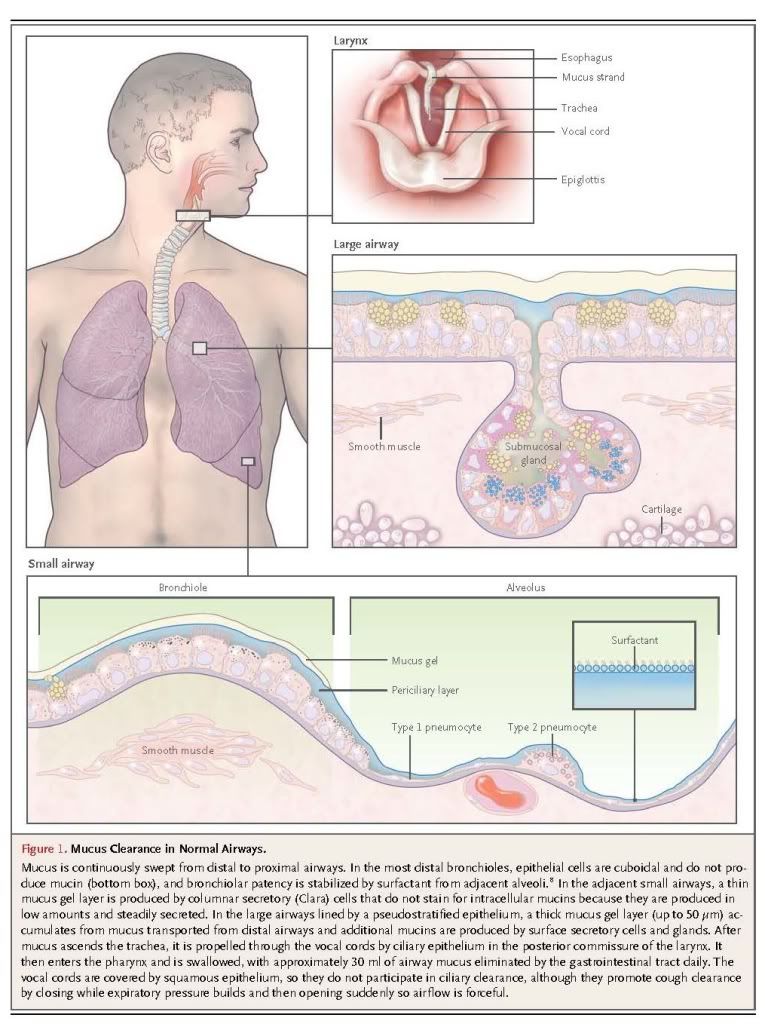
In large airways (luminal diameter, >2 mm), submucosal glands contribute to the secretion of mucins and liquid (Figure 1). Each gland is connected to the airway lumen by a superficial ciliated duct that propels secretions outward and a deeper nonciliated collecting duct.17,18 The body of the gland is located between the spiral bands of smooth muscle and the cartilage plates. Mucous cells constitute approximately 60% of the gland volume, and based on studies in primates, it has been estimated that half as much intracellular mucin is stored in submucosal glands as is stored in surface epithelial cells.19 Serous cells, located distally, make up the remaining approximately 40% of the gland and secrete proteoglycans and numerous antimicrobial proteins. In pathologic states, the volume of submucosal glands can increase to several times the normal volume.20,21
Mucus Gel Layer
A gel is a dilute network that holds shape; thus, although it is composed mostly of liquid, it has many physical characteristics of a solid. Mucus is a gel with properties of both a soft (deformable), elastic solid and a viscous fluid.1,4,5,22,23 Normal mucus is 97% water and 3% solids (mucins, nonmucin proteins, salts, lipids, and cellular debris). Mucins, exceedingly large glycoproteins (up to 3×106 D per monomer) with regions rich in serine and threonine residues linked by their hydroxyl side groups to sugar chains (O-glycosylation), account for less than 30% of the solids.3,4,6,15,24Mucins are 50 to 90% carbohydrate, and they are highly anionic because most of their terminal sugars contain carboxyl or sulfate groups. There are 17 genes encoding mucins in the human genome, of which the gene products of seven are secreted and the remainder is membrane-bound.3,4,6 Five of the secreted mucins have terminal cysteine-rich domains that can form disulfide bonds resulting in polymers that impart the properties of a gel (Figure 2). Two of these polymers, MUC5AC and MUC5B, are strongly expressed in the airways and are detected in similar quantities in human mucus.3,4
MUC5AC and MUC5B form homotypic polymers (i.e., MUC5AC monomers bond only with MUC5AC, and MUC5B monomers bond only with MUC5B), structured as long single chains rather than branches (Figure 2). They form the mucus gel both by entanglement in a mesh and by noncovalent calcium-dependent cross-linking of adjacent polymers.1,3 The glycan side chains bind large amounts of liquid (hundreds of times their weight), which allows mucus to act as a lubricant and the gel layer to serve as a liquid reservoir for the periciliary layer.2 The hydration of mucus dramatically affects its viscous and elastic properties, which in turn determine how effectively it is cleared by ciliary action and cough.1-5,22 Healthy mucus contains 3% solids, with the consistency of egg white. However, mucin hypersecretion or dysregulation of surface liquid volume may increase the concentration of solids up to 15%, resulting in viscous and elastic mucus that is not easily cleared.1,5,22,, In addition, dehydrated mucus adheres more readily to the airway wall. 23,25
Since infection is often initiated by the recognition of host epithelial surfaces by microbial sugar-binding proteins, mucin glycans help sequester pathogens by providing a diverse “glycoprotein landscape” for interaction with these microbial proteins, and patterns of glycosylation can change during inflammation.3,5,26 In addition, the mucus gel layer acts as a solid physical barrier to most pathogens.1,3,5,7 However, the pore size of the gel mesh is sufficiently large (approximately 500 nm) that it is readily penetrated by small viruses with hydrophilic capsids; this has implications for microbial infection and gene therapy.5
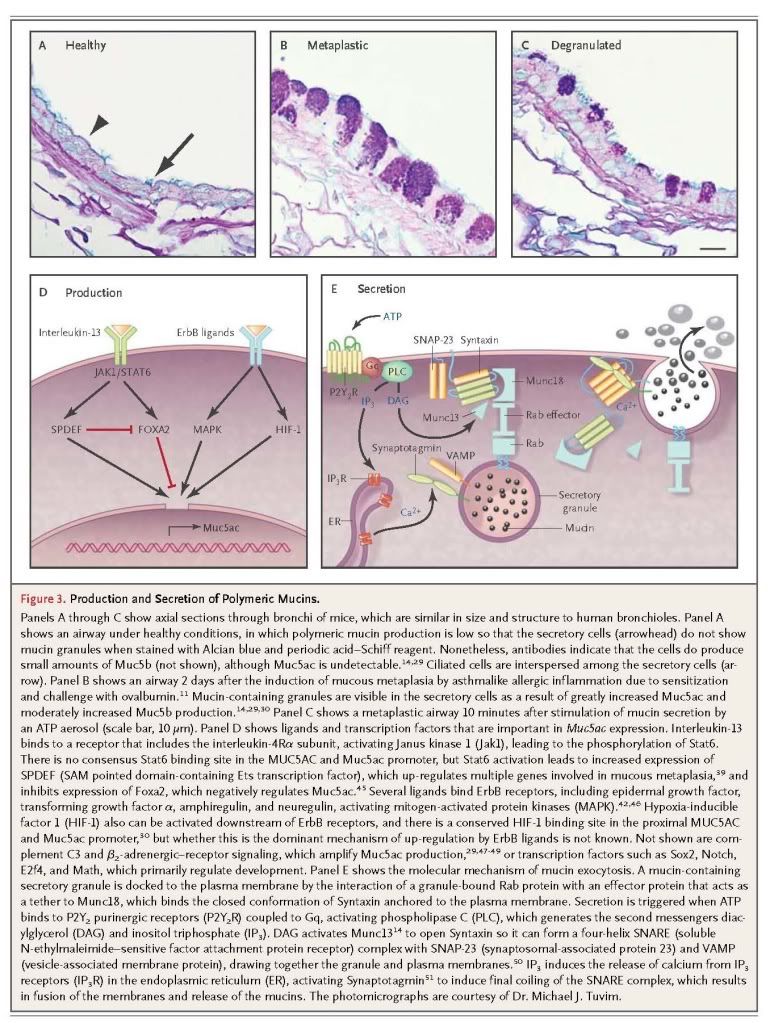
Many other stimuli that increase MUC5AC and Muc5ac expression, such as viruses,31 the smoke component acrolein,40and the cytokines interleukin-4, 9, 17, 23, and 25,52-54 do so, at least in part, through interleukin-13. Overexpression of proinflammatory cytokine interleukin-1β or interleukin-17 increases Muc5ac expression,32,54 whereas interleukin-6 and tumor necrosis factor α do not themselves increase Muc5ac expression but do so indirectly by augmenting the intensity of allergic inflammation.55,56 The control of MUC5B and Muc5b expression is less well understood.57
Mucin Secretion
The secretion of polymeric mucins is regulated separately from mucin production.50,58 The most important secretagogue for surface epithelium appears to be ATP, which acts on apical membrane P2Y2 receptors.59-61 It is not yet clear whether other agonists such as acetylcholine or histamine directly activate receptors on airway epithelial cells or induce airway smooth-muscle contraction leading to ATP release.59,62-64 The continuous presence of low levels of ATP in airway-surface liquid (see below) causes continuous low activity of the secretory machinery, resulting in the steady release of mucins that provide a normal barrier. When mucin production is increased so that mucins accumulate intracellularly (Figure 3B), and secretion of a large number of granules is then triggered (mucus hypersecretion) (Figure 3C), airway luminal occlusion can occur.13,65-67 It might seem that the secretion of a mucin granule would result in no net change in the volume of luminal air space if epithelial-cell volume decreased by the same amount as the volume of secreted mucin. However, mucins are stored in dehydrated form within secretory granules, and they swell to several hundred times their dehydrated volume after secretion as a result of hydration and the exchange of each calcium counterion within the granule for two sodium ions in the extracellular space.9,68 Rapid secretion can deplete airway-surface liquid, resulting in the formation of concentrated, rubbery mucus that is resistant to dilution once the mucin network is formed.1,5,17 Submucosal glands continuously secrete polymeric mucins at a low level and can be further stimulated by adrenergic, cholinergic, and nonadrenergic, noncholinergic nerves.17
Periciliary Layer
The airway mucus gel lies atop a periciliary layer approximately 7 μm deep (Figure 2A). The depth of this layer is critically important for mucociliary clearance (see below). Since the airway epithelium is highly permeable to water, liquid volume is determined by the amount of sodium chloride in the airway lumen.63 In turn, the amount of sodium chloride is regulated primarily by sodium absorption through the epithelial sodium channel and chloride extrusion through the cystic fibrosis transmembrane conductance regulator (CFTR) and calcium-activated chloride channels.63,69 As mucus is propelled proximally, there is net salt and water absorption (>90%) commensurate with the decreasing total cross-sectional area of the airways.2 Locally, the depth of the periciliary layer is fine-tuned by the concentrations of adenine and uridine nucleotides and the metabolite adenosine. Adenine nucleotides are released through channels from ciliated cells that sense mechanical stress during ventilation63,70 and by exocytosis along with uridine nucleotides from secretory cells.60,61 These nucleotides activate P2Y2 receptors and adenosine activates A2b receptors on the apical membrane of ciliated cells, causing changes in intracellular second messengers that promote chloride release and inhibit sodium absorption; as a result, water moves into the airway lumen.60,61
Membrane-bound mucins contribute to the physical properties of liquid near the cell surface, conferring features of a “grafted gel” rather than a fluid on the periciliary layer (Boucher RC, University of North Carolina: personal communication). MUC4 is densely expressed on cilia, configured like parallel bottle brushes, where it prevents penetration by the mucus gel layer and provides lubrication through bound water.4,6 MUC1 is much smaller than MUC4 and is present on the cell surface and microvilli of both ciliated and secretory cells. It has a cytoplasmic tail capable of intracellular signaling, and it modulates pathogen defense and inflammation.6,71 MUC16, the largest mucin, is expressed by both ciliated and secretory cells, and it can be cleaved and incorporated into the mobile gel layer.6,72
Clearance Mechanisms
The mucus gel is propelled in a proximal direction by ciliary beating, clearing inhaled particles, pathogens, and dissolved chemicals that might damage the lungs.2 Polymeric mucins are continuously synthesized and secreted to replenish the gel layer. Normal cilia beat 12 to 15 times per second, resulting in a velocity of the gel layer of approximately 1 mm per minute.73 The rate of mucociliary clearance increases with greater hydration,2,73 and the rate of ciliary beating can be increased by purinergic, adrenergic, cholinergic, and adenosine-receptor agonists,60,73 as well as irritant chemicals.74
A second mechanism for the expulsion of mucus from the airways is cough clearance. This may help explain why lung diseases caused by impaired ciliary function are less severe than those caused by dehydration, which impedes both clearance mechanisms.2 Although cough contributes beneficially to the clearance of mucus in diseases of excessive production or impaired ciliary function, it can also be a troublesome symptom.75,76
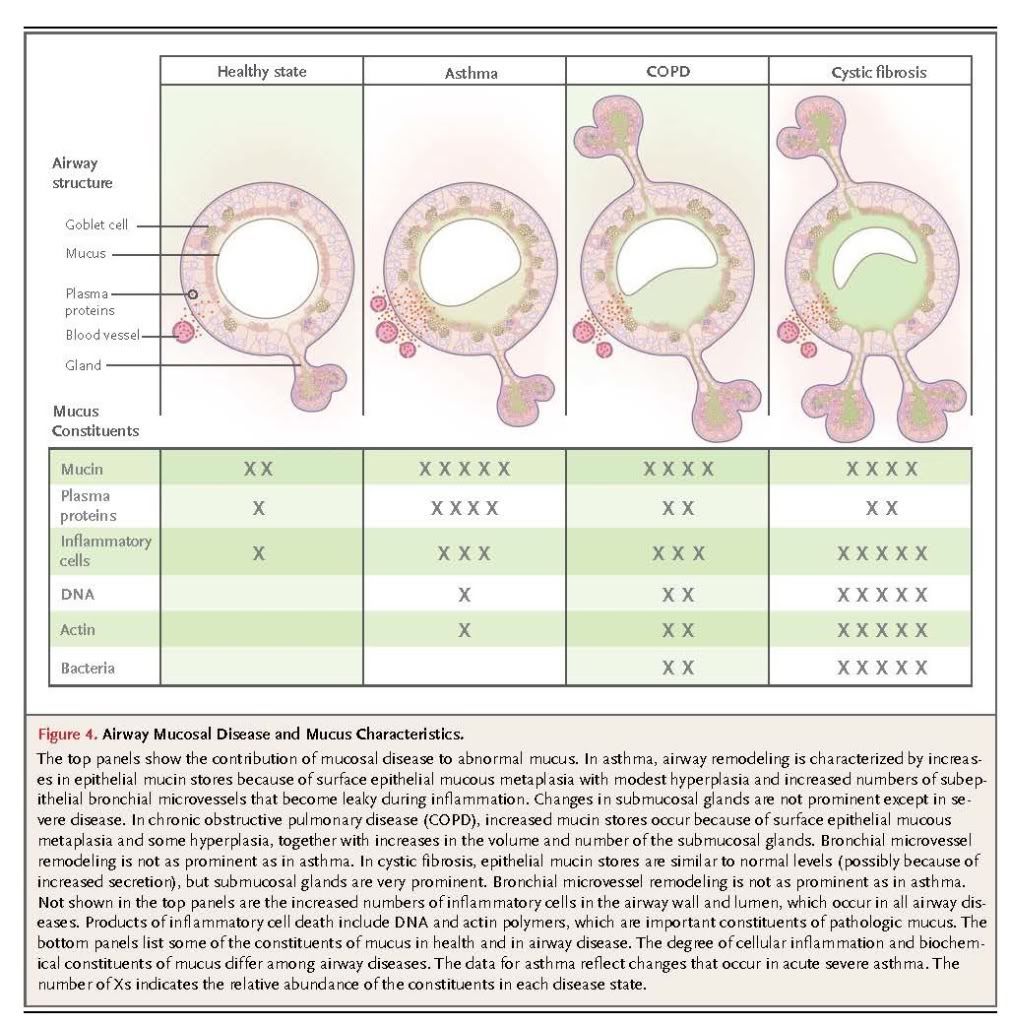
The accumulation of mucus results from some combination of overproduction and decreased clearance, and persistent accumulation can lead to infection and inflammation by providing an environment for microbial growth.
The principal symptoms of impaired mucus clearance are cough and dyspnea. Cough is caused by the stimulation of vagal afferents in the intrapulmonary airways or the larynx and pharynx.75,76 Patients often infer that laryngopharyngeal stimulation, described as “a tickle in the throat,” results from “postnasal drip,” since they recognize that gravity causes mucus to descend from the nasopharynx but are generally unaware that it also ascends from the lungs by ciliary action. Dyspnea is caused when mucus obstructs airflow by occupying the lumen of numerous airways.21,65-67 Physical signs of impaired mucus clearance include cough, bronchial breath sounds, rhonchi, and wheezes. Retained mucus and inflammatory exudates may appear as localized atelectasis or linear or branched opacities on plain radiographs of the chest, and as luminal filling in proximal airways or tree-in-bud opacities in peripheral airways on computed tomographic examination.77 It is important to recognize the role of mucus in clinical presentation. It is necessary to clear mucus from the airway lumen in order to resolve symptoms and allow effective delivery of aerosol therapies. In addition, the presence of mucus may be a sign of underlying inflammation or infection that may warrant additional treatment.
Cystic Fibrosis
Cystic fibrosis is caused by mutations in the gene encoding CFTR, which result in reduced chloride secretion and increased sodium absorption. Together, these result in insufficient airway luminal liquid.78,79 In addition, reduced bicarbonate secretion may result in excessive mucin cross-linking by calcium.80 In transgenic mice with overexpression of a subunit of the epithelial sodium channel in airway epithelial cells, airway luminal liquid is insufficient and a cystic fibrosis–like phenotype develops.81 This finding, coupled with a large amount of supportive data from in vitro studies of human airway epithelial-cell function, has led to a broad consensus that the major consequences of CFTR dysfunction in the airway are dehydration of mucus and reduction in the height of the periciliary layer, particularly in response to infectious or toxic insults.63,69,78,79 These changes result in poor mucus clearance, which sets up a vicious cycle of infection, inflammation, and injury. In patients with cystic fibrosis, mucus has the following characteristics: infiltration with neutrophils and high concentrations of neutrophil-derived DNA and filamentous actin22,82,83; infection with organisms such as Pseudomonas aeruginosa, Staphylococcus aureus, and aspergillus species, often in biofilms at the epithelial-cell surface; and dehydrated, highly entangled polymeric macromolecules that form a gel matrix with a pore size reduced from the normal size of approximately 500 nm to approximately 150 nm.5 Decreased pore size is postulated to immobilize microbes within the mucus gel, thereby promoting biofilm formation, and to inhibit the movement of neutrophils that might otherwise clear the infection.5,69 The net effects of these processes are manifested radiographically as bronchiectasis; pathologically as neutrophilic inflammation, airway fibrosis, and increased numbers of mucin-secreting cells, especially in the submucosal glands; and clinically as cough, purulent sputum, hemoptysis, dyspnea, recurrent lung infections, and rapid loss of lung function.78,79,84
Asthma
The central role of diffuse mucus plugging of the airways in the pathophysiology of asthma has been recognized by pathologists for more than 100 years.65,66,85 However, mucus dysfunction in asthma is often underappreciated by clinicians, possibly because cough in asthma infrequently results in expectoration or because the unavailability of therapies to clear mucus plugs has diverted attention exclusively toward reversing bronchoconstriction and inflammation.86 Mucous metaplasia (i.e., increased surface epithelial mucin production) and an increased number of bronchial microvessels are important components of the airway remodeling in asthma that confers a predisposition to mucus dysfunction.86 These changes occur in patients with airway inflammation characterized by infiltration of the airway wall and luminal mucus with CD4+ T cells, eosinophils, and innate immune cells that secrete Th2 cytokines,85,87 although neutrophil infiltration may also be prominent in acute exacerbations.84 Airway occlusion by mucus plugs can cause localized atelectasis that is evident radiographically in patients with acute asthma exacerbations, and widespread mucus plugging is consistently detected at autopsy in patients with fatal asthma.65,66Diffuse airway narrowing from a combination of concentric smooth-muscle contraction and luminal obstruction with mucus makes asthma uniquely dangerous among airway diseases in its propensity for sudden exacerbations.
Airway mucus in severe asthma has a rubbery quality that contributes to impaired clearance and plug formation. Biochemical analysis of these plugs shows high concentrations of mucins and plasma proteins,84,88 and biophysical analysis shows high entanglement density and elastic modulus.89 Another important pathologic role of plasma proteins in forming these highly elastic mucus plugs is shielding mucins from protease digestion.89 Since neutrophil elastase activity is increased in the airways of patients with asthma in the recovery phase of near-fatal exacerbations, it may help to digest mucus plugs in these patients.90 A history of persistent symptoms related to sputum is associated with more severe disease phenotypes in chronic asthma,91 and mucus hypersecretion is especially problematic in allergic bronchopulmonary aspergillosis.85
Chronic Obstructive Pulmonary Disease
Small-airway mucus obstruction is characteristic of COPD, even in patients who do not expectorate sputum or who have an emphysematous phenotype.92,93 Conversely, patients with COPD who have copious expectoration may have little airflow obstruction, probably because the mucus comes from large airways and causes minimal occlusion. Despite this weak correlation with sputum production, airflow obstruction does correlate with changes in mucin gene expression,38 increases in goblet-cell number and size,38 the occlusion of small airways with mucus,88 and expansion of the submucosal glands.21,92 Mucus dysfunction induced by cigarette smoke is complex and incompletely understood, but it involves adverse effects on the structure and function of cilia,94-96– activation of ErbB receptors,41 decreased function of CFTR,97 and proinflammatory effects that increase mucin production while decreasing mucus hydration and clearance. Cigarette smoke contains multiple toxins, including particulate matter, oxidative chemicals, and organic compounds, among which acrolein is important because it potently induces mucin production. 40
Increased mucin production and decreased luminal liquid in COPD have deleterious consequences for airway health, as they do in asthma and cystic fibrosis, including mucus stasis and airway infection. Haemophilus influenzae, P. aeruginosa, Streptococcus pneumoniae, and Moraxella catarrhalis are detected in sputum in 25 to 50% of adults with COPD. The infection rate increases with increasing disease severity, and the acquisition of new bacterial strains is associated with COPD exacerbations.98 On the basis of studies in a mouse model in which H. influenzae lysate elicited airway inflammation and fibrosis but not mucous metaplasia,56 one may speculate that in COPD, a reduction in mucus clearance that is related to cigarette smoke leads to airway infection, which, in turn, leads to inflammation and fibrosis.
Other Airway Diseases Associated with Mucus Dysfunction
Mucus dysfunction occurs in virtually all inflammatory airway diseases. Acute viral and bacterial infections and chronic diseases such as primary ciliary dyskinesia, non–cystic fibrosis bronchiectasis (which is often caused by atypical mycobacterial infection), panbronchiolitis, and immunodeficiency states (e.g., hypogammaglobulinemia, human immunodeficiency virus infection, organ transplantation, and hematologic malignant conditions) all have a component of mucus dysfunction. In addition, retained mucus is a problem in intubated patients and those in whom lung mechanics are disrupted as a result of paralysis, immobilization, or surgery; atelectasis and pneumonia are common complications in such patients. Genomic markers in chromosomal region 11p15.5 (which encompasses MUC5AC and MUC5B) have been reported to be associated with asthma severity,99 and panbronchiolitis,100 although mechanisms leading to disease susceptibility have not yet been defined.
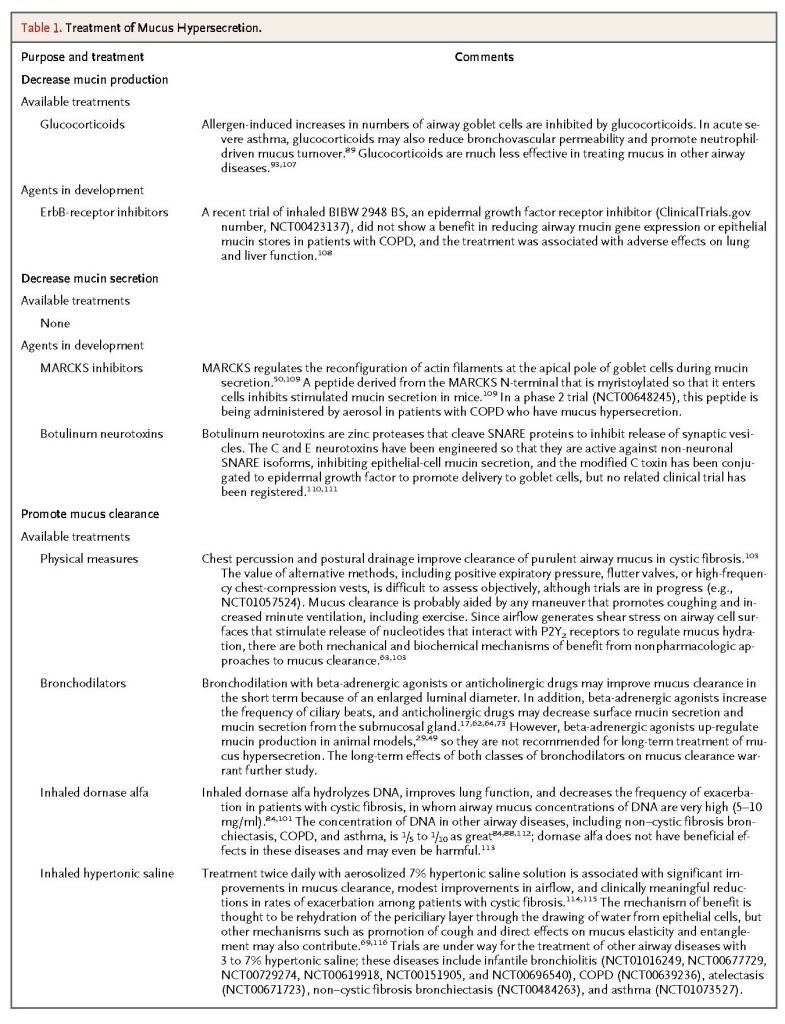
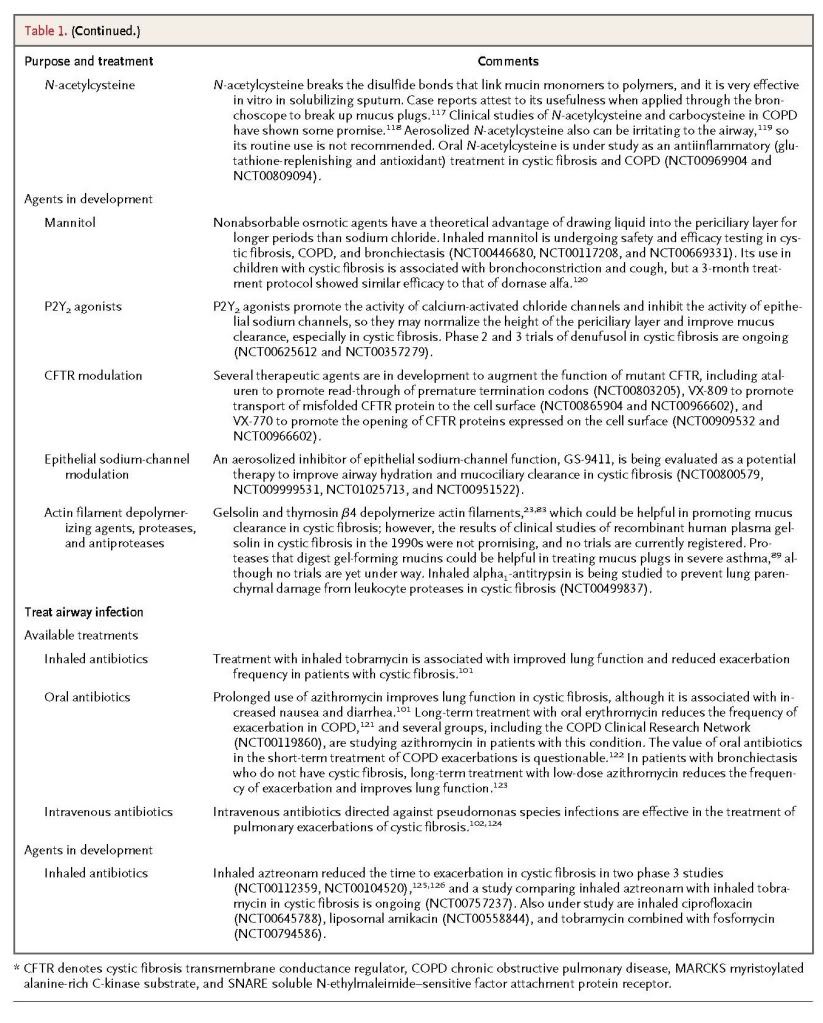
Recent insights into the formation of pathologic mucus in disease have led to the introduction of tailored therapies such as hydration by means of aerosolized hypertonic saline solutions or the reduction of mucus viscosity and elasticity by aerosolized dornase alfa. Targeted treatment of pathologic airway mucus not only improves symptoms of cough and dyspnea but also decreases the frequency of disease-related exacerbations and slows disease progression. Elucidation of how mucin production is controlled is still needed, since that might allow the development of additional therapies to prevent overproduction.


 留言列表
留言列表
 線上藥物查詢
線上藥物查詢 