

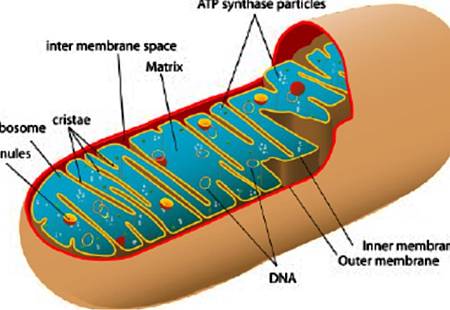
Mitochondria are mobile organelles that exist in dynamic networks. They continuously join by the process of fusion and divide by the process of fission. Mitochondria are derived from eubacterial endosymbionts that are capable of aerobic respiration; this finding was proposed independently by Merezhkovsky in 1905 and by Margulis in 1967.1 First described as “bioblasts” by Altmann in 1890, it was Benda's 1898 observation of their heterogeneous morphologic features, sometimes ball-shaped and other times linear, that inspired the name mitochondrion, from the Greek words mitos (meaning thread) and chondrion (meaning granule).2 Lewis and Lewis's 1914 observations established the field of mitochondrial dynamics. They noted, “Any one type of mitochondria such as a granule, rod or thread may at times change into any other type or may fuse with another mitochondrium [sic], or it may divide into one or several mitochondria.”3The once secret, dynamic lives of mitochondria are revealed by confocal, live-cell imaging with the use of potentiometric dyes or mitochondria-targeted fluorescent proteins (see Fig. 1 in theSupplementary Appendix, available with the full text of this article at NEJM.org).
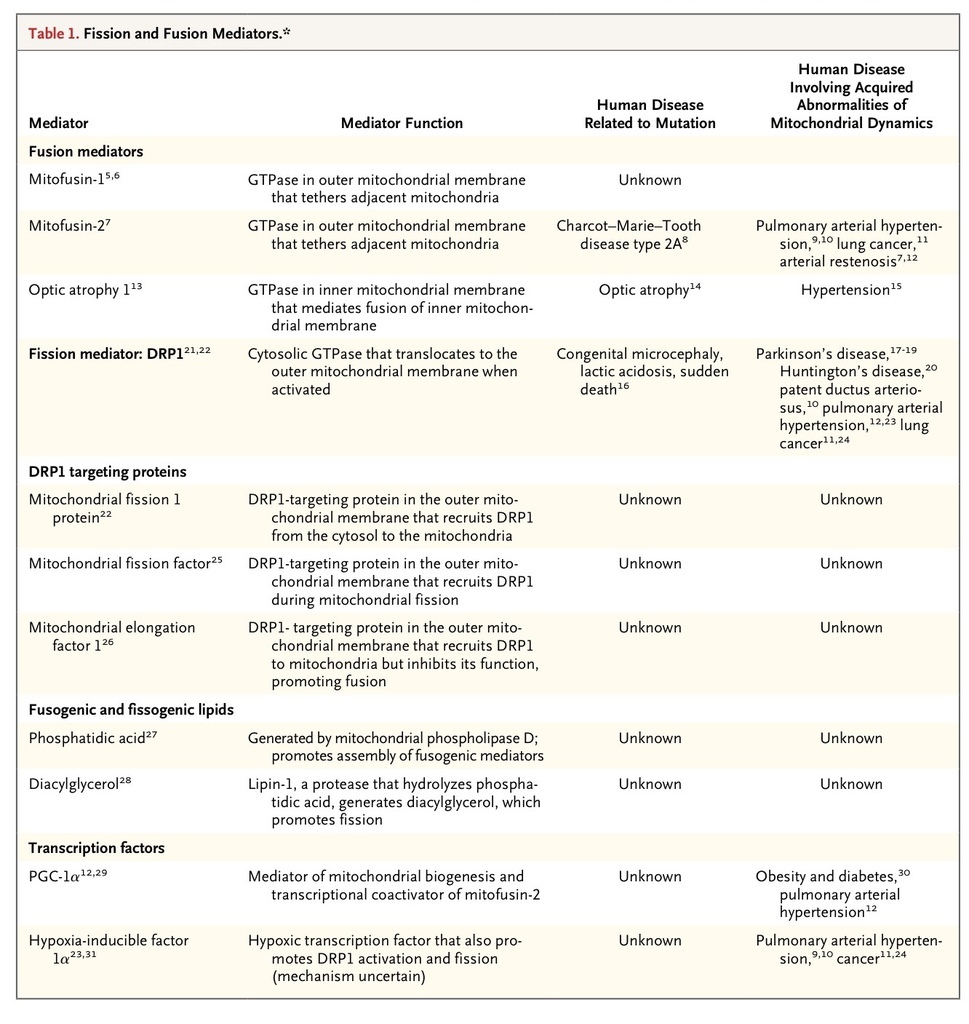
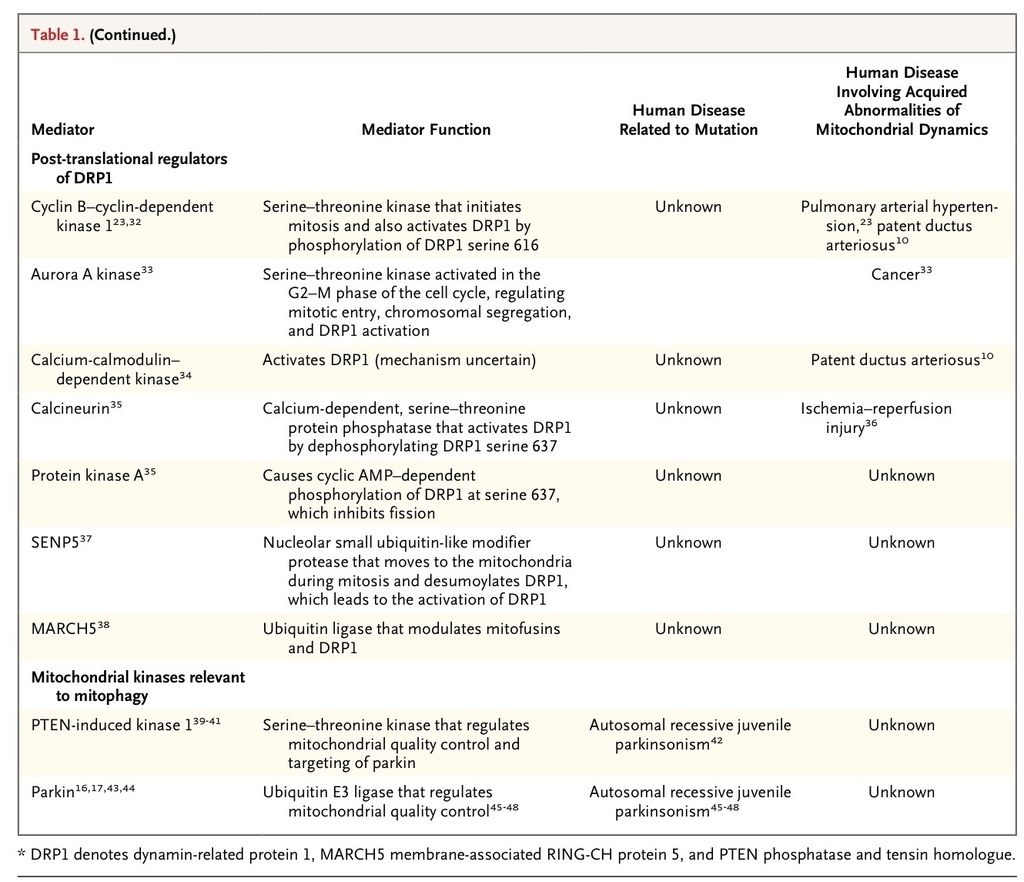
Most mitochondrial proteins, including all those involved in fission and fusion, are nuclear-encoded.4Mutations in 228 nuclear and 13 mitochondrial genes cause rare monogenic syndromes in which mitochondrial dysfunction is unequivocally central to the pathogenesis of the disease. Examples of these syndromes include the MELAS syndrome (mitochondrial encephalomyopathy, lactic acidosis, and strokelike episodes caused by mutation of mitochondrial transfer RNAs), and Leigh's disease (caused by mutations in genes related to oxidative phosphorylation).4 In contrast, disorders of mitochondrial structure are just emerging as mechanisms of disease. Although some disorders of mitochondrial dynamics result from monogenic mutation, most reflect changes in the function or activity of fission and fusion mediators triggered by changes in the cellular milieu. There is an emerging recognition that disordered mitochondrial dynamics contribute to the pathogenesis of complex diseases that are not classically considered to involve mitochondria; these diseases include cancer, cardiovascular disease, and neurodegenerative diseases. Recent identification of the molecular mediators of mitochondrial dynamics (Table 1
) and recognition of their post-translational regulation by an extensive network of kinases, phosphatases, and ubiquitination mediators offer a new understanding of cell biology and novel therapeutic targets. Fission and fusion fine-tune fundamental cellular processes such as calcium homeostasis and the generation of ATP and reactive oxygen species and consequently have important roles in cell-cycle progression, apoptosis, mitophagy, and oxygen sensing.
Although ATP generation is the primary function of the mitochondria (Figure 1FIGURE 1 Mitochondrial Metabolism and Dynamics.), much of the impact of mitochondrial dynamics relates to effects of structure on the nonbioenergetic capabilities of the organelle. Five noncanonical mitochondrial capabilities are centrally involved in the form–function dynamic of the mitochondria (Figure 2FIGURE 2
Mitochondrial Metabolism and Dynamics.), much of the impact of mitochondrial dynamics relates to effects of structure on the nonbioenergetic capabilities of the organelle. Five noncanonical mitochondrial capabilities are centrally involved in the form–function dynamic of the mitochondria (Figure 2FIGURE 2 Mitochondrial Fusion and Fission.). These relationships are bidirectional, meaning they both alter and are altered by mitochondrial dynamics. First, mitochondria are linked to the endoplasmic reticulum at specialized regions of membrane adherence called mitochondria-associated membranes, which facilitate calcium flux into the mitochondria. Mitochondria-associated endoplasmic reticulum membranes allow localized increases in calcium levels to propagate throughout a cell in wavelike patterns.49 This endoplasmic reticulum–mitochondria connection has implications for calcium homeostasis and metabolism. Close coupling of these organelles increases mitochondrial calcium levels, which may initiate apoptosis50 or, at physiologic levels, enhance oxidative metabolism by activating pyruvate dehydrogenase.51 Mitochondria–endoplasmic reticulum connectivity is regulated by mitofusin-2 and can create microdomains that facilitate fission9,27 (Figure 2B, inset).
Mitochondrial Fusion and Fission.). These relationships are bidirectional, meaning they both alter and are altered by mitochondrial dynamics. First, mitochondria are linked to the endoplasmic reticulum at specialized regions of membrane adherence called mitochondria-associated membranes, which facilitate calcium flux into the mitochondria. Mitochondria-associated endoplasmic reticulum membranes allow localized increases in calcium levels to propagate throughout a cell in wavelike patterns.49 This endoplasmic reticulum–mitochondria connection has implications for calcium homeostasis and metabolism. Close coupling of these organelles increases mitochondrial calcium levels, which may initiate apoptosis50 or, at physiologic levels, enhance oxidative metabolism by activating pyruvate dehydrogenase.51 Mitochondria–endoplasmic reticulum connectivity is regulated by mitofusin-2 and can create microdomains that facilitate fission9,27 (Figure 2B, inset).
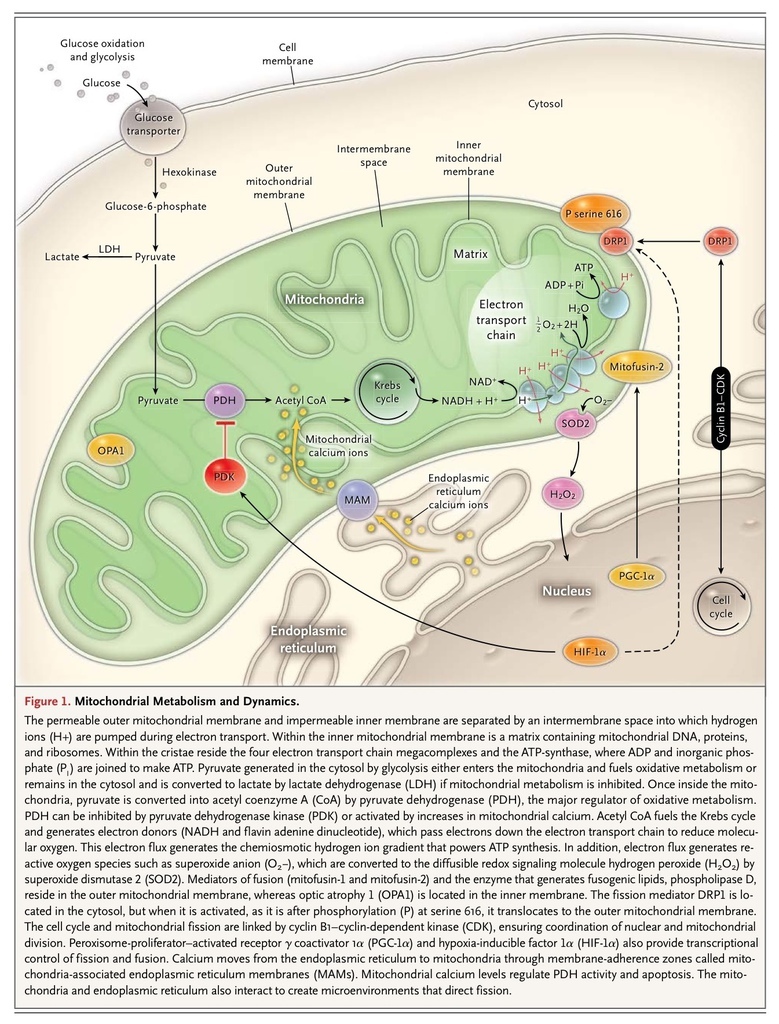

Second, mitochondrial numbers are regulated by mitochondrial biogenesis to meet the energy demands of the cell and compensate for cell damage. This process is mediated by peroxisome-proliferator–activated receptor γ coactivator 1α (PGC-1α), which is relevant to mitochondrial dynamics since it is a transcriptional coactivator of the fusion mediator mitofusin-2 (Table 1).12,29
Third, mitochondria have a quality-control program called mitophagy. Mitophagy maintains cellular health by selectively enclosing damaged, depolarized mitochondria in autophagic vacuoles for elimination by lysosomes (Figure 3A
).52,53 Fission isolates depolarized mitochondria while coordinated down-regulation of fusion mediators prevents network reintegration, thereby facilitating mitophagy.
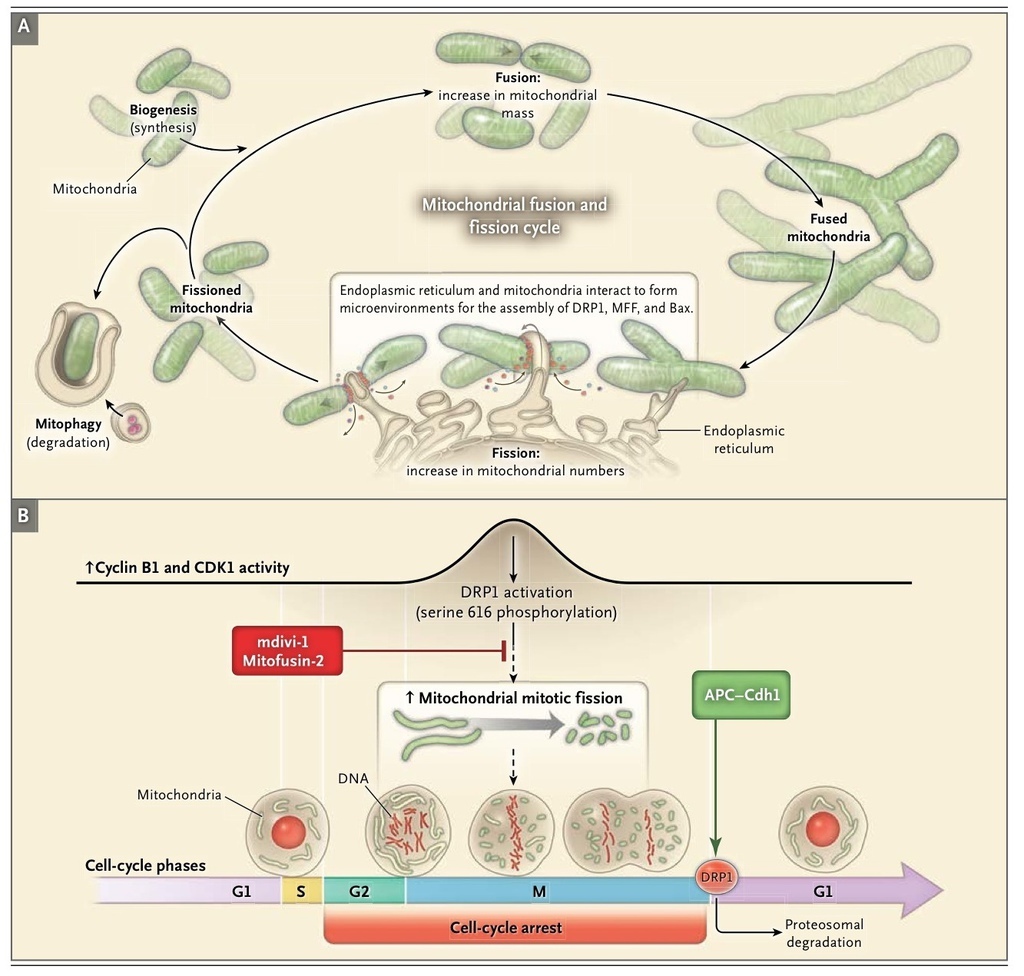

Fourth, mitochondria actively traverse the cytosol on dynein and kinesin tracks.54 It is uncertain whether mobility and mitochondrial dynamics have an obligatory relationship; however, dynein, a molecular motor for mitochondrial transport, also regulates fission.55
Fifth, mitochondria are oxygen sensors in cells within the homeostatic oxygen-sensing system, such as pulmonary arterial and ductus arteriosus smooth-muscle cells.56 These specialized mitochondria vary production of diffusible reactive oxygen species by the electron transport chain in proportion to cellular oxygen levels, permitting redox regulation of ion channels, enzymes, and transcription factors.56 Mitochondrial dynamics are an early step in this redox signaling mechanism.10,23
Fission creates smaller, more discrete mitochondria, which, depending on the context, are more capable of generating reactive oxygen species, facilitating mitophagy, or accelerating cell proliferation. Fusion results in a more interconnected mitochondrial network that enhances communication with the endoplasmic reticulum. Fusion also allows diffusion of matrix content among mitochondria (Figure 2A), diluting the accumulated mitochondrial DNA mutations57 and oxidized proteins. Both fission and fusion are mediated by a small number of highly conserved, guanosine triphosphatases (GTPases)58,59 (Figure 2 and Table 1). Fission is mediated by dynamin-related protein 1 (DRP1),60,61 a cytosolic protein that on activation translocates to the outer mitochondrial membrane. Here, DRP1 multimerizes, creating a ringlike structure that constricts and divides the organelle21,22 (Figure 2B). Video 1, available at NEJM.org, shows the dynamic nature of fission and fusion when a photoactivated green fluorescent protein–labeled mitochondrion divides (fission) and then fuses with a red fluorescent protein–labeled mitochondrion to create a fused yellow mitochondrion. DRP1 is actively targeted to the outer mitochondrial membrane by non-GTPase receptor proteins such as mitochondrial fission protein 1 (Fis1),22 mitochondrial fission factor (MFF),25 and mitochondrial elongation factor 126 (Figure 2B and Table 1). Assembly of the fission apparatus is assisted by the endoplasmic reticulum, which contacts the mitochondria, creating a microdomain for assembly of DRP1, MFF, and proapoptotic proteins62 (Figure 2B andFigure 3A).
DRP1 activity is rapidly regulated by the opposing effects of phosphorylation at two key serines. Phosphorylation of serine 616 increases DRP1 activity, whereas phosphorylation of serine 637 decreases it. Each serine is targeted by different kinases and phosphatases, thereby linking mitochondrial fission to crucial cellular processes (Figure 2B, inset; and Table 1). For example, phosphorylation of serine 616 by the mitosis initiator, cyclin B1–cyclin-dependent kinase (cyclin B1–CDK1), activates DRP1 coordinating fission to cell division23,32 (Figure 3B), whereas phosphorylation by calcium-calmodulin–dependent kinase (CamK) links fission to intracellular calcium.34 Phosphorylation of serine 637 by protein kinase A inactivates DRP163; conversely, dephosphorylation of serine 637 by the calcium-sensitive protein phosphatase, calcineurin, activates DRP1 (Figure 2B and Table 1). The ratio of serine 616 to serine 637 phosphorylation determines DRP1 activity and reflects the aggregate effects of many kinases and phosphatases (Figure 2). DRP1 activity is also post-translationally regulated by the ubiquitin ligase membrane–associated RING-CH protein 5 (MARCH5)38 and by small ubiquitin-like modifier type 1 (SUMO1)37 (Table 1and Figure 2B). Our understanding of the role of DRP1 and fission in disease states has been advanced by the availability of mitochondrial division inhibitor 1 (mdivi-1), a small molecule that inhibits the GTPase activity of DRP1, prevents multimerization, and inhibits fission.64
Fusion is mediated by mitofusin-1 and mitofusin-2 isoforms in the outer mitochondrial membrane and by optic atrophy 1 (OPA1) protein in the inner mitochondrial membrane13 (Table 1 and Figure 1A). Mitofusins are targeted to the mitochondria by sequences in their transmembrane and C-terminal domains.65 With cytosolic amino and carboxyl termini, mitofusins initiate fusion by creating homodimeric or heterodimeric, antiparallel, coiled-coil linkages between adjacent mitochondria5(Figure 2A, inset). Mitofusin-2 is also located in the endoplasmic reticulum, where it alters morphologic features and promotes endoplasmic reticulum–mitochondrial tethering,65 thereby enhancing mitochondrial calcium uptake.9 OPA1 has eight splice variants, each with differential fusion activity and mitochondrial protease susceptibility.6 The fusion mediators also regulate mitochondrial metabolism, and when they are down-regulated or dysfunctional, there is generally a reduction in mitochondrial oxidative capacity.6 Fusion and fission are guided by lipids generated by mitochondrial phospholipase D, notably phosphatidic acid.27 The small, negatively charged lipid head group of phosphatidic acid causes negative curvature of lipid bilayers and recruits adaptor proteins, promoting fusion.27 However, phosphatidic acid can be hydrolyzed by lipin-1, creating diacylglycerol, which promotes fission.28
DISEASES INVOLVING MITOCHONDRIAL DYNAMICS
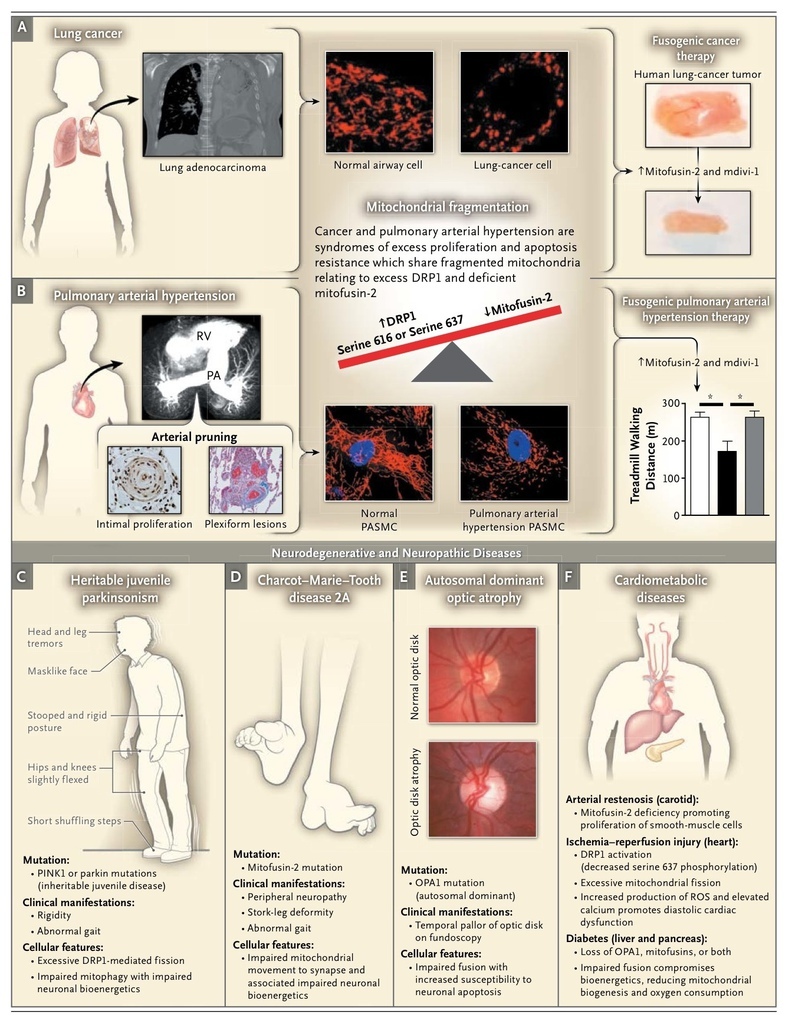
Disorders of mitochondrial dynamics are implicated in neurodegenerative, neoplastic, endocrine, and cardiovascular diseases. The therapeutic implications of the mitochondrial dynamics pathway for these diseases are reviewed in Figure 4
.

Proliferative, Apoptosis-Resistant Diseases
Cancer
Excessive proliferation and resistance to apoptosis are hallmarks of cancer cells (Figure 4A).69 These hallmarks reflect, in part, acquired abnormalities of mitochondrial function, notably a shift from oxidative metabolism to aerobic glycolysis (the Warburg effect).70 This metabolic phenotype primarily reflects activation of mitochondrial pyruvate dehydrogenase kinase, which inhibits pyruvate dehydrogenase and suppresses oxidative metabolism, rendering the cell apoptosis-resistant (Figure 1). These functional mitochondrial abnormalities offer potential therapeutic targets. Restoring pyruvate dehydrogenase activity and oxidative metabolism inhibits proliferation, induces apoptosis, and shrinks human tumors in xenotransplantation models41 and in patients.71 Structural mitochondrial abnormalities also contribute to the imbalance between proliferation and apoptosis in cancer.11 Impaired fusion and enhanced fission fragment the mitochondrial network in lung adenocarcinoma in humans11(Figure 4A).
The role of mitochondrial dynamics in cancer relates to the requirement for mitochondrial division during mitosis. This coordination between mitochondrial division and mitosis (so-called mitotic fission) ensures equitable distribution of mitochondria to daughter cells32,43 (Figure 3B). The molecular basis for the coordination of mitochondrial and nuclear division is emerging. At the transition from the G1 phase to the S phase, mitochondria fuse and increase ATP production.43DRP1 inhibition induces mitochondrial hyperfusion and triggers DNA replication and cyclin E accumulation.43 The coordination of fission and mitosis is substantially regulated by cyclin B1–CDK1, which simultaneously initiates mitosis and activates DRP1 by phosphorylating serine 61611(Figure 2B). Another mitotic kinase, aurora A, phosphorylates the Raslike GTPase (RalA), leading to mitotic, mitochondrial accumulation of RalA and its effector, ralA binding protein 1 (RalBP1). RalBP1 serves as a scaffold for recruiting DRP1 and cyclin-CDK to mitochondria and inducing fission.33
Increased fission in lung-cancer cells and tumors from patients who have not received treatment reflects post-translational DRP1 activation, manifested by an increased ratio of serine 616 to serine 637 phosphorylation.11 In patients with lung cancer, increased DRP1 expression predicts a likelihood of recurrence that is increased by a factor of 3.4, as well as a greater likelihood of cisplatin resistance.24 Impaired fusion, resulting from down-regulation of mitofusin-2, also contributes to network fragmentation.11 Mitochondrial fragmentation contributes to the cancer phenotype in several ways. Fragmentation may accelerate mitotic fission11 and also interrupts intramitochondrial calcium waves, preventing calcium-mediated apoptosis.50 Inhibition of DRP1 or augmentation of mitofusin-2 decreases proliferation and increases apoptosis in cancer cells and leads to regression of human lung tumors in a xenotransplantation model11 (Figure 4A).
Pulmonary Arterial Hypertension
Pulmonary arterial hypertension (Figure 4B) is an obstructive pulmonary vasculopathy. Although vasoconstriction, inflammation, and thrombosis contribute to the pathogenesis, an “oncologic view” is emerging, which holds that excessive proliferation and impaired apoptosis mediate disease progression.17 Many factors contribute to the neoplastic phenotype of pulmonary arterial hypertension.18,72 Both oxygen-sensing and mitochondrial dynamics are disordered in pulmonary-artery smooth-muscle cells, as indicated by normoxic activation of hypoxia-inducible factor 1α (HIF-1α) and mitochondrial fragmentation.19 The fingerprint of mitochondrial dynamics in pulmonary arterial hypertension is similar to that in cancer, with reduced mitofusin-2–mediated fusion12 and excessive DRP1-mediated fission.23
These acquired defects reflect both transcriptional and post-translational abnormalities. Transcriptional abnormalities include increased HIF-1α activation23,31 and decreased PGC-1α activity.12 Activation of HIF-1α in normal pulmonary-artery smooth-muscle cells is sufficient to cause DRP1-dependent mitochondrial fission.23 The normoxic HIF-1α activation in pulmonary arterial hypertension leads to a proliferative, apoptosis-resistant milieu through both metabolic effects (pyruvate dehydrogenase inhibition) and induction of a fission–fusion imbalance; the latter mechanism is not well understood.17
Post-translational abnormalities (increased cyclin B1–CDK1–dependent and CamK-dependent DRP1 phosphorylation) activate DRP1 and promote fission.23 Many of these pro-fission, antifusion abnormalities occur in models of pulmonary arterial hypertension created in healthy rats and persist in cell culture, suggesting epigenetic underpinnings.17
Mitofusin-2 levels are reduced in pulmonary arterial hypertension, contributing to both mitochondrial fragmentation and the proliferative diathesis.12 Mitofusin-2 was originally named the “hyperplasia-suppressor gene”7 because of its antiproliferative effects on systemic arterial smooth-muscle cells.7Mitofusin-2 down-regulation in pulmonary arterial hypertension is associated with low levels of PGC-1α,12 the transcriptional coactivator of mitofusin-2.73 Preclinical testing showed that augmenting mitofusin-2 decreases proliferation and increases apoptosis of pulmonary-artery smooth-muscle cells and leads to partial regression of experimentally induced pulmonary arterial hypertension in vivo in rats.12 Although this therapeutic benefit is associated with restoration of fusion, the link between fusion and inhibition of proliferation is controversial. In systemic arterial cells, truncated mitofusin-2 variants that do not localize to mitochondria retain antiproliferative properties by inhibiting extracellular signal-regulated and mitogen-activated protein kinases.7
Patent Ductus Arteriosus
Functional ductus arteriosus closure is initiated by oxygen-dependent vasoconstriction within minutes after birth. An increase in oxygen is accompanied by increases in oxidative metabolism, electron transport chain activity, and generation of reactive oxygen species. Reactive oxygen species are converted by mitochondrial superoxide dismutase 2 (SOD2) to hydrogen peroxide, a diffusible oxidant messenger that regulates ion channels and enzymes, thereby initiating ductal constriction.56 A subsequent fibrogenic, proliferative mechanism results in anatomical closure.
Recently, it was discovered that the redox-sensing mechanism of the human ductus arteriosus relies on mitochondrial fission. Within 5 minutes after an increase in oxygen, DRP1-mediated fission fragments ductus arteriosus smooth-muscle cell mitochondria.10 Inhibiting DRP1 selectively prevents oxygen-induced ductal constriction. Oxygen induces fission by cyclin B1–CDK1–dependent and CamK-dependent, post-translational DRP1 activation.10 In ductus arteriosus smooth-muscle cells, fission activates pyruvate dehydrogenase and increases electron transport chain complex 1 activity, creating an oxidant environment. Conversely, inhibiting fission interrupts redox signaling, preventing oxygen-dependent production of mitochondrial-derived hydrogen peroxide. This link between structure and electron transport chain assembly and activity is similar to an observation made in drosophila, in which a knockout of PINK1 (phosphatase and tensin homologue–induced putative kinase 1) impairs fission and reduces bioenergetic capacity by causing defective electron transport chain complex assembly.74 Although sustained DRP1 inhibition prevents anatomical closure in an ex vivo human ductus arteriosus model, it is not known whether impaired mitochondrial fission contributes to spontaneous patent ductus arteriosus.
Neurodegenerative Diseases
Neuronal development requires both DRP175 and mitofusin-2.76 Neuronal DRP1 knockout causes mitochondrial maldistribution, apoptosis, and perinatal death due to brain hypoplasia (Figure 4C, 4D, and 4E).75 The beneficial role of DRP1-induced fission in human brain development is highlighted by the description of an infant with a dominant-negative DRP1 mutation.16 She had hypotonia, microcephaly, and optic atrophy and died suddenly at 37 days of age; studies showed impaired mitochondrial metabolism and hyperfusion, which were consistent with impaired fission.16Although some level of fission is required for brain development, increased fission and impaired fusion, resulting in mitochondrial fragmentation, are the rule in adult neurodegenerative diseases.
Familial Parkinsonism
Parkinson's disease is a neurodegenerative syndrome characterized by resting tremor, rigidity, and bradykinesia, resulting from death of dopaminergic neurons in the substantia nigra. Although most cases of parkinsonism are sporadic, rare heritable, juvenile cases result from autosomal recessive mutations in mitochondrial-targeted kinases PINK158 and parkin59 (Table 1 and Figure 4C).
PINK1, a serine–threonine kinase, protects neurons by preventing stress-induced mitochondrial depolarization and apoptosis42 and reducing mitochondrial oxidative stress and fission.39 PINK1 also phosphorylates parkin, targeting it to depolarized mitochondria and thereby enhancing mitophagy.77 Parkin, a ubiquitin E3 ligase, protects neurons by ubiquitinating proteins that are abnormal in structure or quantity (including DRP146), marking them for proteosomal degradation. Parkin also ubiquitinates dysfunctional mitochondria, leading to their removal by mitophagy.78PINK1 and parkin loss-of-function mutations have complex effects; however, increased DRP1-mediated mitochondrial fission is an important consequence and contributes to neuronal death.46,79,80 Neurons lacking PINK1 or parkin accumulate DRP1, and the resulting excessive fission increases oxidative stress and reduces ATP production.39,40,81 These neurons can be rescued by inhibiting fission with the use of mdivi-182 or by enhancing fusion through augmentation of mitofusin-2 or OPA1.39,40,81 Excessive fission is not neuronally restricted; it is detected in dermal fibroblasts from patients.41 How this relatively generalized abnormality of mitochondrial dynamics results in tissue-restricted disease is an unresolved mystery that is relevant to optic atrophy and Charcot–Marie–Tooth disease type 2A.
PINK1 and parkin also regulate mitophagy. The relationship between mitochondrial dynamics and mitophagy is uncertain; however, the autophagic machinery both delivers mitochondria to lysosomes and contributes to mitochondrial fragmentation39 (Figure 3A). At physiologic levels, both mitochondrial fission and mitophagy are beneficial, working together to eliminate damaged and depolarized mitochondria39; however, an excess of either process leads to cell death.
Two pesticides that promote parkinsonism (the electron transport chain complex 1 inhibitor rotenone83 and paraquat84) induce mitochondrial fragmentation. Rotenone and paraquat initiate parkin-mediated mitochondrial depolarization. They then trigger fragmentation by promoting mitofusin degradation, through a proteosomal mechanism mediated by the AAA+ superfamily of ATPases and valosin-containing protein p97.47 The AAA-family ATPases interact with p97 to compose a ubiquitin–proteosome pathway that extracts proteins such as mitofusin-2 from the outer mitochondrial membrane into the cytosol, where they ultimately undergo proteosomal degradation. Removing mitofusins from the outer mitochondrial membrane inhibits fusion and prevents abnormal mitochondria from reintegrating into the network, thereby facilitating mitophagy.47 This so-called outer mitochondrial membrane–associated degradation pathway47 mediates mitochondrial quality control and is analogous to the endoplasmic reticulum quality-control pathway (ERAD).85
Alzheimer's Disease
The most common cause of dementia is Alzheimer's disease, which is characterized by accumulation of amyloid plaques, neurofibrillary tangles, and loss of connections between neurons. In one study, β-amyloid increased neuronal nitric oxide, generating S-nitrosylated, activated DRP1, which caused fission and neuronal damage.86 Inhibition of DRP1 nitrosylation reduced neurotoxicity. However, a subsequent investigation could neither replicate the effects of S-nitrosylation on DRP1 activity nor support a role for this mechanism in Alzheimer's disease.87
Huntington's Disease
An autosomal dominant disease characterized by choreoathetosis, dementia, and premature death, Huntington's disease results from expansion of a CAG trinucleotide DNA sequence encoding a polyglutamine tract in the huntingtin protein.88 The polyglutamine accumulation causes protein misfolding and promotes abnormal protein interactions. Mutant huntingtin interacts with and activates DRP1, increasing fission and neuronal sensitivity to apoptosis in rat neurons and in fibroblasts from patients with Huntington's disease.20 The severity of CAG expansion in the huntingtin gene, an established predictor of disease severity, also predicts the severity of fission.88The importance of polyglutamine-containing proteins for fission and cell death is recapitulated in cellular and Caenorhabditis elegans models of Huntington's disease.88 In both models, inhibiting DRP1 or augmenting mitofusin-2 restores mitochondrial fusion, preserves ATP, and prevents cell death.88
Amyotrophic Lateral Sclerosis
Amyotrophic lateral sclerosis is a motor neuron disease that causes weakness and death. In a rare heritable form of the disease, mutation of superoxide dismutase 1 causes DRP1-mediated fission, which inhibits axonal transport of mitochondria, causing neuronal death.89 The mitochondrial matrix sirtuin SIRT3 and PGC-1α provide protection against excessive fission and neuronal death.
Neuropathies
Charcot–Marie–Tooth Disease
An inherited peripheral neuropathy, Charcot–Marie–Tooth disease (Figure 4D) usually results in atrophy of calves, foot deformities, and abnormal gait. The axonal subtype of Charcot–Marie–Tooth disease, type 2A, is generally due to autosomal dominant, missense mutations in the fusion-mediating, coiled-coil, and GTPase domains of mitofusin-2.8,90 Mitofusin-2 mutations inhibit metabolism, causing energetic deprivation that limits mitochondrial mobility to synapses.61 A single mitofusin-2 mutation can result in a heterogeneous disease phenotype and an onset of disease at various ages within one family.90
Optic Atrophy
Mutations in OPA1 cause an autosomal dominantly inherited optic neuropathy14 (Figure 4E). Affected patients have progressive loss of visual acuity that often leads to blindness by the second decade of life.91 The loss of OPA1-mediated fusion leaves fission unopposed; this predisposes retinal ganglion cells to apoptosis, ultimately resulting in optic-nerve degeneration.91 Most of the approximately 96 known mutations occur in the GTPase domain of OPA1 or its 3′ terminus, resulting in dominant-negative and haploinsufficiency phenotypes, respectively.67 A mitofusin-2 mutation can also result in optic atrophy, a reminder of the interdependence of these fusion pathways.92
Cardiometabolic Diseases
Diabetes Mellitus
Mitochondrial fusion defects promote diabetes in murine models of type 1 diabetes mellitus. In patients with obesity or type 2 diabetes mellitus (Figure 4F), fusion is also impaired, as evidenced by reduced mitofusin-2 expression and decreased mitochondrial size.30 Hepatic deletion of mitofusin-2 increases reactive oxygen species generation, impairs insulin signaling, and causes glucose intolerance.93 Down-regulation of mitofusin-2 impairs oxygen consumption and promotes diabetes. Mice with pancreatic β-cell–specific OPA1 knockout also have impaired glucose-stimulated insulin secretion, and hyperglycemia develops in such mice.94 Although severe impairment of fusion promotes diabetes, tonic restoration of fusion is unlikely to be a simple remedy because some mitochondrial fragmentation is required for mitophagy. Mitochondria undergo asymmetric fission, resulting in both normal mitochondria and depolarized, dysfunctional mitochondria that are deficient in OPA1.95 OPA1 deficiency isolates abnormal mitochondria, preventing network reintegration. This localized impairment of fusion and mitophagy is beneficial. Indeed, inhibiting mitophagy by enhancing fusion (overexpressing OPA1) leads to accumulation of damaged mitochondria with impaired respiration and reduced insulin secretion.95
Ischemia–Reperfusion Injury
Despite effective resuscitation techniques, mortality from cardiac arrest remains high, in part because of ischemia–reperfusion injury (Figure 4F). In the kidney, mdivi-1 protects renal tubular cells from fission induced by ischemia–reperfusion injury96; it is also protective in a cardiac ischemia–reperfusion injury model.97 During cardiac arrest, DRP1 is activated by calcineurin-mediated dephosphorylation of DRP1 at serine 637.36 The resulting fission increases reactive oxygen species production, increases levels of calcium, and impairs diastolic relaxation. DRP1 inhibition, whether achieved directly or by targeting calcineurin, may have a therapeutic effect (Figure 4F).
Cardiomyopathy
Because germline mitofusin knockouts are lethal at the embryonic stage, tissue-specific, conditional knockout mice are required to study the role of mitofusins in the heart. Conditional cardiac ablation of both mitofusin isoforms simultaneously has deleterious effects on mitochondrial morphologic features, respiration, and contractile function, resulting in death from cardiac failure.98,99Conversely, deletion of only one mitofusin isoform is cardioprotective against ischemia–reperfusion injury and reactive oxygen species. Conditional cardiac deletion of mitofusin-2 results in mitochondrial dysfunction and hypertrophy, both of which are mild, with a slight reduction in left ventricular function. However, these mice have reduced susceptibility to activation of the mitochondrial permeability transition pore.100 The same group of investigators subsequently showed that conditional mitofusin-1 knockouts also have normal baseline cardiac function and are protected from reactive oxygen species–induced cardiotoxicity.
FUTURE DIRECTIONS
Mitochondrial dynamics are involved in the mechanisms of a variety of human diseases and may offer therapeutic targets. Before attempts are made to manipulate mitochondrial dynamics therapeutically, further evaluation is required to identify the optimal molecular targets and define safe and effective doses of fission and fusion modulators that selectively target the relevant cellular populations. Additional pharmacologic or molecular modulators of fission and fusion are needed. Qi et al.101 recently described a small, rationally designed peptide inhibitor (P110) that prevents DRP1 activation and fission by interfering with the protein–protein interaction between DRP1 and its mitochondrial adaptor target protein, Fis1. Validation of the use of peripheral cells (i.e., fibroblasts) to assess fission and fusion and metabolism is required. The usefulness of measuring mediators of mitochondrial dynamics in peripheral blood as disease biomarkers merits study. Future research directions are highlighted in Table 1 in the Supplementary Appendix.




 留言列表
留言列表
 線上藥物查詢
線上藥物查詢 