

N Engl J Med 2014; 370:847-859February 27, 2014DOI: 10.1056/NEJMra1208626
The definition of coagulopathy is “a condition in which the blood's ability to clot is impaired.” However, for some clinicians, the term also covers thrombotic states, and because of the complexity of the hemostatic pathways, the two conditions can exist simultaneously. Some practitioners would consider that mildly abnormal results on coagulation screening without bleeding can also indicate a coagulopathy. This review is confined to the original definition of coagulopathy as given above. Such states are common in patients in the intensive care unit (ICU) and require a clinicopathological approach to ensure that the correct diagnosis is made and the appropriate treatment administered. The lack of evidence for managing coagulopathies in critical care is striking. This review will highlight selected areas in which there is high-quality evidence and at the same time point out areas for which there is poor evidence. In the latter case, there is little consensus on management.
DIFFERENTIAL DIAGNOSIS
A medical history taking and physical examination are vital, since many different conditions can produce similar laboratory abnormalities. For example, end-stage liver failure and disseminated intravascular coagulation produce thrombocytopenia and similar changes in standard tests of coagulation, and yet the management of and prognosis for these conditions are very different. A peripheral-blood smear is a vital investigation tool in most cases to confirm a low platelet count and the presence or absence of other diagnostic features, such as red-cell fragmentation, platelet morphologic abnormalities, or evidence of dysplasia or hematinic deficiency. Table 1
TABLE 1
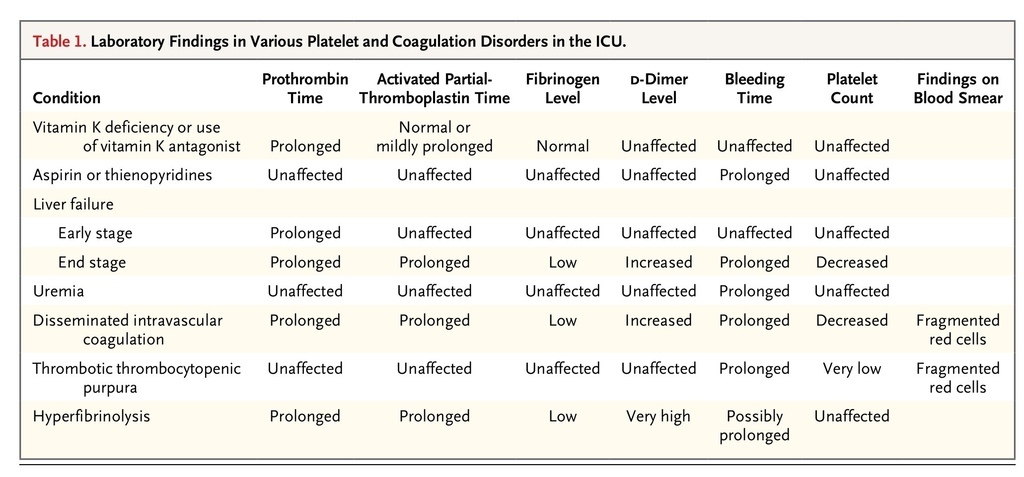
Laboratory Findings in Various Platelet and Coagulation Disorders in the ICU.
and Figure 1
FIGURE 1
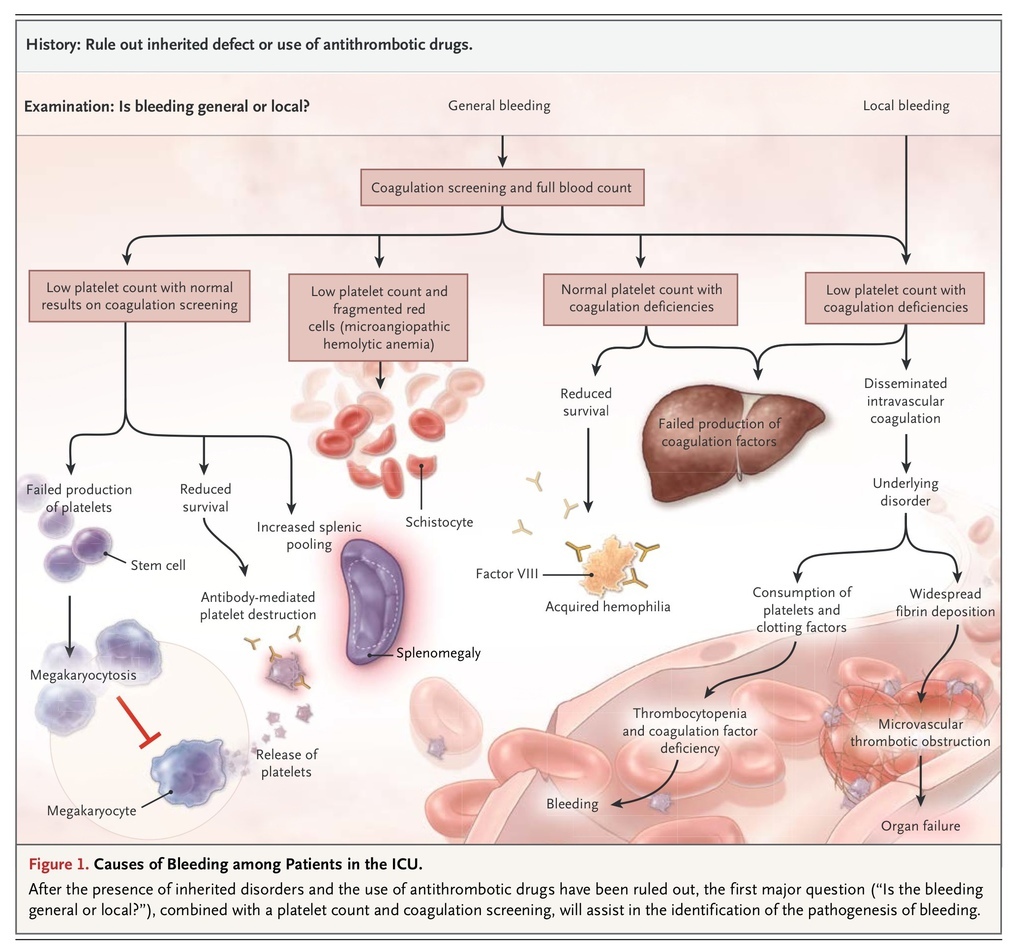
Causes of Bleeding among Patients in the ICU.
highlight the relationship between laboratory findings and various coagulopathies.
Once it has been determined that the underlying cause is not a response to therapeutic agents meant to modify the coagulation response (e.g., treatment with vitamin K antagonists, heparinoids, or direct factor Xa or IIa inhibitors), practitioners need to evaluate the pattern of bleeding, which may include widespread petechiae and mucosal bleeding in platelet disorders, generalized oozing from de-epithelialized surfaces, and fast bleeding from damaged major vessels.
MANAGEMENT OF COAGULOPATHIES
The first principle of the management of coagulopathies in critical care is to avoid the correction of laboratory abnormalities with blood products unless there is a clinical bleeding problem, a surgical procedure is required, or both.
Major Bleeding
The lack of good-quality evidence is most marked in the use of blood components to manage major bleeding. When blood components were introduced into critical care practice decades ago, their benefit was never assessed in randomized clinical trials. Later, concern about transfusion-transmitted infections (human immunodeficiency virus infection, hepatitis, and a new variant of Creutzfeldt–Jakob disease) and limitations in the blood supply led to a more restrictive use of blood components.
In the absence of randomized, controlled trials, retrospective studies of military casualties1 and, later, similar studies of civilian casualties2 showing improved survival with transfusion of 1 U of fresh-frozen plasma for each unit of red cells have resulted in earlier administration of an increased number of units of fresh-frozen plasma. However, these studies have been criticized, particularly for methodologic flaws that include survival bias (i.e., patients who did not survive were not transfused with fresh-frozen plasma) and heterogeneity between studies.3
Despite the lack of evidence that bleeding after surgery and gastrointestinal or obstetric hemorrhage are associated with hemostatic changes similar to those in acute traumatic coagulopathy, the early use of a transfusion ratio of fresh-frozen plasma to red cells of 1:1 or 1:2 has become widespread. This increased use of plasma is not risk-free, since the incidence of transfusion-related acute lung injury is increased,4 as may be the risk of the acute respiratory distress syndrome (ARDS). In one study involving trauma patients requiring a nonmassive transfusion (<10 U of packed red cells within 12 hours after admission), the administration of more than 6 units of fresh-frozen plasma, as compared with no transfusion, was associated with an increase by a factor of 12 in the rate of ARDS and an increase by a factor of 6 in the multiple organ dysfunction syndrome.5
The critical transfusion ratio of fresh-frozen plasma to red cells in the management of major bleeding is not known. This question is being evaluated in the North American Pragmatic, Randomized Optimal Platelets and Plasma Ratios study (ClinicalTrials.gov number, NCT01545232). This multicenter, randomized trial is comparing the effect of various ratios of blood products administered to trauma patients who are predicted to require massive transfusion (>10 U of packed red cells within the next 24 hours) on rates of death at 24 hours and 30 days. In the interim, a North American–European divide in the practice of using blood components to support hemostasis has emerged. Although in North America there has been increased use of fresh-frozen plasma in patients with major hemorrhage, some European practitioners have abandoned the use of fresh-frozen plasma, relying on the exclusive use of factor concentrates on the basis of rotational-elastometry–guided intervention with prothrombin complex concentrate, factor XIII, and fibrinogen.6 In contrast, other practitioners believe that the treatment of major hemorrhage should begin with fibrinogen supplementation with tranexamic acid, a synthetic derivative of the amino acid lysine that acts as an antifibrinolytic agent by competitively inhibiting plasminogen, with red cells and intravenous fluid used on an as-needed basis.7
Fibrinogen is a critical molecule in coagulation. It is the protein that ultimately forms fibrin, the ligand for platelet aggregation, and in patients with major bleeding, it is required to a larger extent than any other hemostatic protein.8 In such patients, this requirement reflects increased consumption, loss, dilution, and fibrinogenolysis. On the basis of these multiple roles, even in the absence of evidence from randomized, controlled trials, guidelines for the management of traumatic bleeding now indicate that the trigger level for supplementing fibrinogen should be 1.5 to 2.0 g per liter rather than 1.0 g per liter.9 It is unknown whether early fibrinogen supplementation and the use of prothrombin complex concentrate, as compared with the use of fresh-frozen plasma, improves clinical outcomes in patients with major bleeding. Future randomized, controlled trials should assess the overall benefit and safety, including the rate of hospital-acquired venous thromboembolism.10,11 Similarly, the use of recombinant factor VIIa, which has been shown to reduce the use of red cells in bleeding but not to reduce mortality, needs further evaluation. Data from placebo-controlled trials have shown that the off-label use of recombinant factor VIIa significantly increased the risk of arterial thrombosis.12,13
Tranexamic acid should be administered to all patients with major bleeding after trauma. This recommendation is supported by a large, randomized, controlled trial, the Clinical Randomization of an Antifibrinolytic in Significant Hemorrhage (CRASH-2) study, in which 20,000 trauma patients with bleeding or at risk for major bleeding were randomly assigned to receive either tranexamic acid or placebo. Patients who received tranexamic acid within 3 hours after injury had a one-third reduction in deaths from bleeding.14 After a secondary analysis of their data, the CRASH-2 investigators recommended that tranexamic acid be administered as soon as possible after injury, since the drug ceased to confer benefit and appeared to be associated with increased mortality if it was administered more than 3 hours after injury.15 Reassuringly for a hemostatic drug, the incidence of thrombosis after trauma was not increased in the study patients. Strong evidence that tranexamic acid reduced the need for blood transfusion in surgery has been available for years, although the effect of tranexamic acid on thromboembolic events and mortality in such patients remains uncertain. 16
HEMOSTATIC SUPPORT FOR INVASIVE PROCEDURES
There is no supportive evidence for the prophylactic use of fresh-frozen plasma to correct abnormal results on coagulation screening (for prothrombin time, activated partial-thromboplastin time, and fibrinogen) before an invasive procedure. Coagulation screening has no predictive value for later bleeding, and the use of fresh-frozen plasma may not correct abnormal results on coagulation tests. There is currently no consensus on what coagulation-screening results should trigger the use of fresh-frozen plasma, which has led to variation in the use of fresh-frozen plasma by critical care physicians.17,18 Thrombin generation, although delayed, is normal or enhanced19 when the prothrombin ratio is 1.5 or less, so I suggest that a prothrombin ratio of 1.5 or less is satisfactory for the insertion of a central venous or an arterial catheter in patients in whom direct compression can assist with hemostasis without the need for prophylactic supplementation with fresh-frozen plasma.
As a general rule, dietary intake of vitamin K, which is necessary for the formation of coagulation factors II, VII, IX, and X, may be inadequate in critical care settings.20 Despite the lack of high-quality evidence and the inability of vitamin K to correct a coagulopathy caused by liver disease, I recommend supplementation of vitamin K1 (at a dose of at least 1 mg orally daily or 10 mg intravenously weekly) for critical care patients at risk.
DISSEMINATED INTRAVASCULAR COAGULATION
Disseminated intravascular coagulation is a clinicopathological diagnosis21 of a disorder that is defined by the International Society on Thrombosis and Hemostasis (ISTH) as “an acquired syndrome characterized by the intravascular activation of coagulation with loss of localization arising from different causes.” This condition typically originates in the microvasculature and can cause damage of such severity that it leads to organ dysfunction (Figure 2
FIGURE 2

Pathogenesis of Disseminated Intravascular Coagulation in Sepsis.
). It can be identified on the basis of a scoring system developed by the ISTH (Table 2
TABLE 2
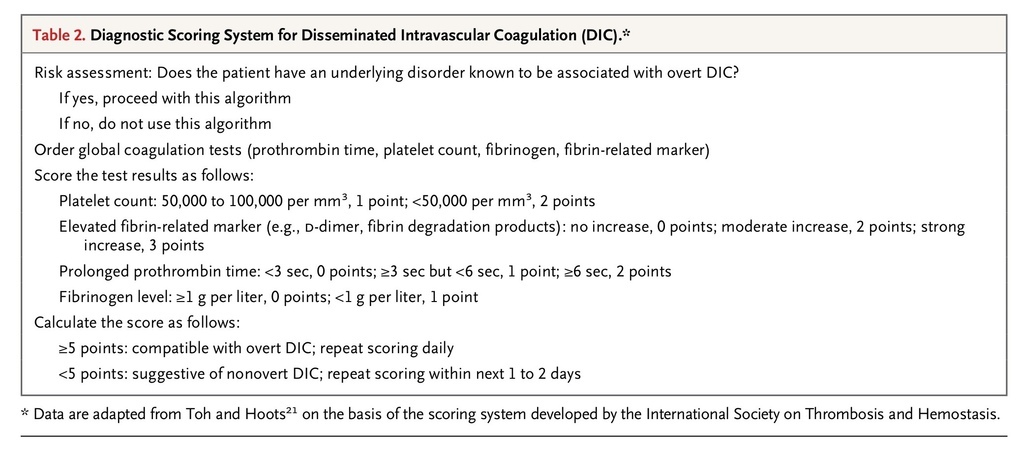
Diagnostic Scoring System for Disseminated Intravascular Coagulation (DIC).
).
Disseminated intravascular coagulation usually presents as hemorrhage, with only 5 to 10% of cases presenting with microthrombi (e.g., digital ischemia) alone. Whether the condition presents with a thrombotic or bleeding episode depends on its cause and host defenses. Sepsis is the most common cause of disseminated intravascular coagulation in critical care; systemic infection with a range of bacteria from Staphylococcus aureus to Escherichia coli is known to be associated with this condition. The complex pathophysiology is mediated by pathogen-associated molecular patterns, which generate an inflammatory response in the host through signaling at specific receptors. For example, signaling by means of toll-like and complement receptors initiates intracellular signaling, which results in the synthesis of several proteins (including proinflammatory cytokines). These proteins trigger hemostatic changes, leading to the up-regulation of tissue factor22 and impairment of physiologic anticoagulants and fibrinolysis. Tissue factor plays a critical role in this process, as shown in meningococcal septicemia, in which the level of tissue factor on monocytes at presentation may be predictive of survival.23 Another study of meningococcal sepsis showed that a large amount of tissue factor was found on monocyte-derived circulating microparticles.24 The up-regulation of tissue factor activates coagulation, leading to the widespread deposition of fibrin and to microvascular thromboses and may contribute to multiple organ dysfunction.
Complex abnormalities of the physiologic anticoagulants occur, and pharmacologic doses of activated protein C, antithrombin, and tissue factor pathway inhibitor appeared to be beneficial in a study of endotoxemia in animals. These promising studies led to major randomized, controlled trials of the supplementation of physiologic anticoagulants with pharmacologic doses of activated protein C,25 antithrombin,26 and tissue factor pathway inhibitor27 in patients with sepsis. However, the studies showed no reductions in the rates of death and increased bleeding episodes.
The consumption of the coagulation proteins and platelets produces a bleeding tendency, with thrombocytopenia, a prolonged prothrombin time and activated partial-thromboplastin time, hypofibrinogenemia, and elevated levels of fibrin degradation products, such as D-dimers. The physiologic anticoagulants are also consumed in the process of inhibiting the many activated coagulation factors.19 In fulminant disseminated intravascular coagulation, the consumption and diminished supply of platelets and coagulation proteins usually results in oozing at vascular access sites and wounds but occasionally causes profuse hemorrhage.
The cornerstone for managing this condition remains the management of the underlying cause (e.g., sepsis). Further management may not be necessary in patients with mild abnormalities in coagulation and no evidence of bleeding. Guidelines for management are based mainly on expert opinion, which suggests replacement of coagulation proteins and platelets in patients who are bleeding. Platelet transfusion is indicated to maintain a platelet level of more than 50,000 per cubic millimeter, along with the administration of fresh-frozen plasma to maintain a prothrombin time and activated partial-thromboplastin time of less than 1.5 times the normal control time and a source of fibrinogen to maintain a fibrinogen level of more than 1.5 g per liter. 28
The use of antifibrinolytic agents is contraindicated in the management of disseminated intravascular coagulation, since the fibrinolytic system is required in recovery to ensure the dissolution of the widespread fibrin. Some guidelines recommend the administration of therapeutic doses of unfractionated heparin in patients with a thrombotic phenotype (e.g., gangrene),28 but such recommendations remain controversial because of the difficulties in monitoring treatment in a patient who already has a prolonged activated partial-thromboplastin time; in addition, heparin administration may provoke hemorrhage. Currently, there is insufficient clinical evidence to make a firm recommendation on the use of heparin in patients with disseminated intravascular coagulation.
THROMBOCYTOPENIA
Pathophysiological Mechanisms
Thrombocytopenia may arise because of decreased production or increased destruction (immune or nonimmune) of platelets, as well as from sequestration in the spleen. Among patients who are admitted to an ICU, the condition occurs in about 20% of medical patients and a third of surgical patients. The cause of this condition is often multifactorial. Patients with thrombocytopenia tend to be sicker, with higher illness-severity scores, than those who are admitted with normal platelet counts.29 Table 3
TABLE 3

Differential Diagnosis of Thrombocytopenia in the ICU.
lists the differential diagnoses of thrombocytopenia in the critical care setting. Given the long list, it is important to identify patients in whom thrombocytopenia requires specific and urgent action (e.g., heparin-induced thrombocytopenia and thrombotic thrombocytopenic purpura). Drug-induced thrombocytopenia is a diagnostic challenge, because critically ill patients often receive multiple medications that can cause thrombocytopenia.
A platelet threshold of 10,000 per cubic millimeter for platelet transfusion in patients who are in stable condition is both hemostatically efficacious and cost-effective in reducing platelet-transfusion requirements.30 Patients with sustained failure of platelet production, such as those with myelodysplasia or aplastic anemia, may remain free of serious hemorrhage, with counts below 5000 to 10,000 per cubic millimeter. However, a higher platelet transfusion trigger should be set in patients with other hemostatic abnormalities or increased pressure on platelet turnover or platelet function. If the patient is actively bleeding, then a platelet count of 50,000 per cubic millimeter should be maintained. Among patients who have or are at risk for bleeding in the central nervous system or who are undergoing neurosurgery, a platelet count of more than 100,000 per cubic millimeter is often recommended, although data are lacking to support this recommendation.29,30
Standard platelet counts are produced by cell counters that categorize the cells according to size, but large platelets may be the same size as red cells and thus be categorized as such. Therefore, an immunologic method of platelet counting, in which platelet antigens are labeled with markers that can be detected with the use of flow cytometry, may be helpful in providing a true count.31 Since platelet transfusions may lead to immune platelet refractoriness owing to the formation of anti-HLA antibodies, the use of HLA-matched platelets, if available, should produce better platelet counts after transfusion.
Immunologic Causes
As a general rule, an abrupt reduction in platelet counts with a history of recent surgery suggests an immunologic cause or adverse transfusion reaction (post-transfusion purpura or drug-induced thrombocytopenia). Heparin-induced thrombocytopenic thrombosis is an uncommon, transient, drug-induced, autoimmune prothrombotic disorder caused by the formation of IgG antibodies that cause platelet activation by the formation of antibodies to complexes of platelet factor 4 and heparin.32
Post-Transfusion Purpura
Post-transfusion purpura is a rare bleeding disorder caused by a platelet-specific alloantibody (usually, anti–human platelet antigen 1a [HPA-1a]) in the recipient. HPA-1a reacts with donor platelets, destroying them and also the recipient's own platelets. The majority of affected patients are multiparous women who have been sensitized during pregnancy. Treatments for post-transfusion purpura include intravenous immune (gamma) globulin, glucocorticoids, and plasmapheresis. High-dose intravenous immune globulin (2 g per kilogram of body weight administered over either 2 or 5 days) produces an increased platelet count in about 85% of patients. Large numbers of platelet transfusions may be required to control severe bleeding before there is a response to intravenous immune globulin. There is limited evidence that the use of HPA-1a–negative platelets is more effective than the use of platelets from random donors.33
Thrombotic Microangiopathies
Profound thrombocytopenia and microangiopathic hemolytic anemia (red-cell fragmentation) characterize the thrombotic microangiopathies, which includes three major disorders: thrombotic thrombocytopenic purpura, the hemolytic–uremic syndrome, and the HELLP syndrome (characterized by pregnancy-related hemolysis, elevated liver-enzyme levels, and low platelet count). The majority of cases of thrombotic thrombocytopenic purpura are due to a deficiency of a disintegrin and metalloproteinase with thrombospondin type 1 motif 13 (ADAMTS13), a disorder that may be hereditary or caused by autoimmune destruction. The absence of ADAMTS13 results in the persistence of high-molecular-weight von Willebrand factor, which can cause spontaneous platelet aggregation when the protein is subjected to high shear stress. The rate of death in untreated cases is nearly 95%, but with early plasmapheresis, the survival rate is 80 to 90%. The use of rituximab, a chimeric monoclonal antibody against the surface B-cell protein CD20, which leads to destruction of those cells, has been shown to reduce the rate of recurrence of the autoimmune form of this disorder from 57% to 10%.34 Thrombotic thrombocytopenic purpura is a medical emergency and in untreated cases is associated with a rate of death of 90%, usually from myocardial infarction due to platelet thrombi in the coronary arteries. Thus, an active diagnosis of this disorder or failure to rule it out should lead to urgent plasmapheresis.
LIVER DISEASE
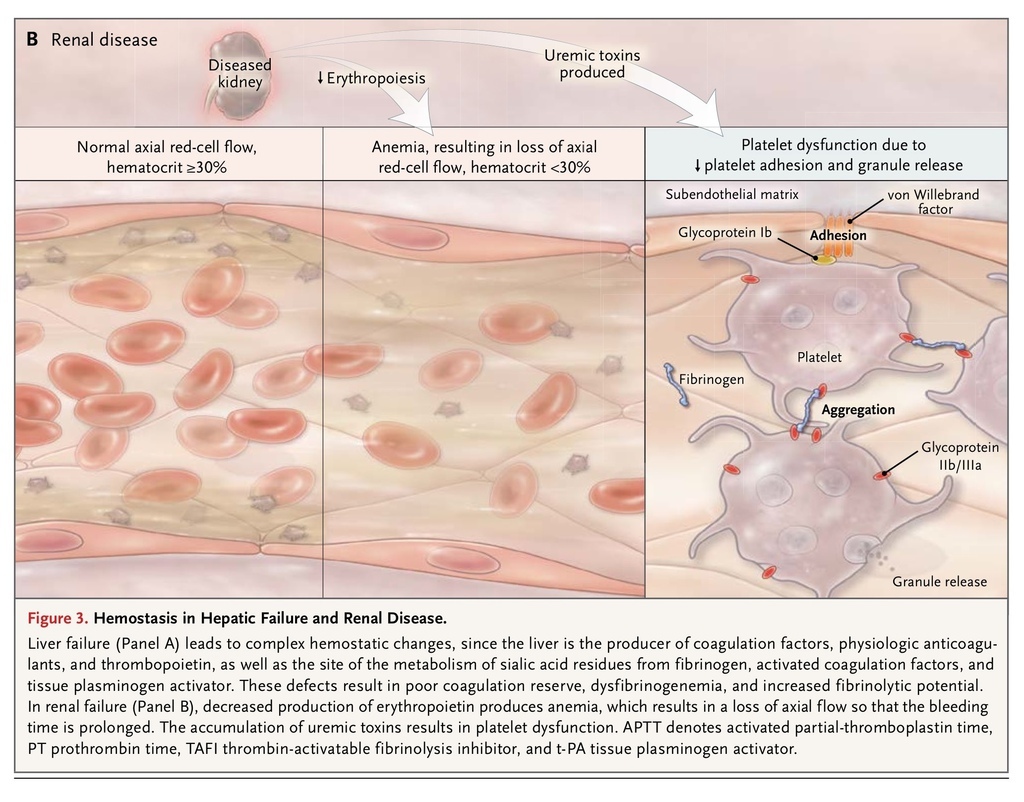
Thrombopoietin and most hemostatic proteins are synthesized in the liver. Thus, reduced hepatic synthetic function results in prolongation of the screening tests of coagulation (particularly the prothrombin time) and reduced platelet counts, although levels of factor VIII and von Willebrand factor are increased.35 Acute alcohol intake inhibits platelet aggregation. In chronic liver disease, there is also increased fibrinolytic potential due to the failure of the liver to metabolize tissue plasminogen activator. In cholestatic liver disease, there is reduced absorption of lipid-soluble vitamins, so reduced amounts of the vitamin K–dependent coagulation factors II, VII, IX, and X are produced. Furthermore, in liver disease, the failure of the normal enzymatic removal of sialic acid from fibrinogen results in dysfibrinogenemia36 (Figure 3A
FIGURE 3
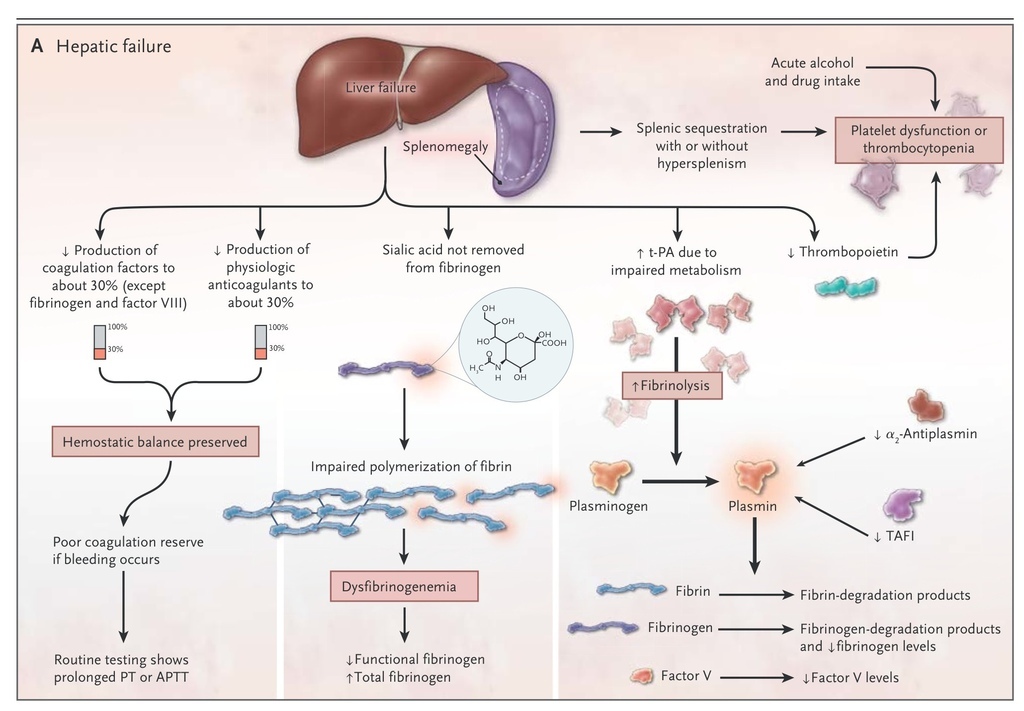
Hemostasis in Hepatic Failure and Renal Disease.
).
However, in parallel with the reduction in coagulation factors, there is a similar reduction in the production of physiologic anticoagulants. Thus, patients with chronic liver disease and a prolonged prothrombin time are no longer considered to have a deficiency of coagulation factors, since their coagulation is rebalanced and thrombin generation is usually normal.35 In such cases, there is no need to treat prolonged coagulation times in the absence of bleeding. If bleeding does occur in liver disease, then consensus guidelines, which are largely based on consensus expert opinion, recommend blood-component management as determined by the results of testing of the platelet count, prothrombin time, activated partial-thromboplastin time, thrombin time, and fibrinogen. In a recent randomized, controlled trial, investigators compared a liberal red-cell transfusion strategy (hemoglobin level, <9 g per deciliter) and a restrictive strategy (hemoglobin level, <7 g per deciliter) in patients with acute upper gastrointestinal bleeding. Patients who were treated with the restrictive strategy had longer survival (6 weeks) and a lower rate of rebleeding than did those who were treated with the liberal strategy.37 In this study, portal circulation pressures increased significantly among patients in the liberal-strategy group. Although there are no similar studies addressing changes in coagulopathy or thrombocytopenia, it seems sensible to adopt a restrictive approach to the use of fresh-frozen plasma and platelets in patients with acute upper gastrointestinal bleeding. The role of tranexamic acid in patients with gastrointestinal bleeding is under investigation in the ongoing randomized, controlled Hemorrhage Alleviation with Tranexamic Acid–Intestinal System (HALT-IT) trial (NCT01658124). In patients with liver disease and laboratory tests indicating abnormal synthesis of coagulation factors, vitamin K should be routinely administered to aid in the synthesis of coagulation factors.
RENAL DISEASE
Uremic bleeding typically presents with ecchymoses, purpura, epistaxis, and bleeding from puncture sites due to impaired platelet function. The platelet dysfunction is a result of complex changes that include dysfunctional von Willebrand factor, decreased production of thromboxane, increased levels of cyclic AMP and cyclic GMP, uremic toxins, anemia, and altered platelet granules, all of which are necessary for adequate formation of a platelet plug (Figure 3B). The anemia that commonly accompanies renal disease leads to the loss of laminar flow in arterioles so that red cells no longer push platelets and plasma to the endothelium, leading to prolongation of the bleeding time; treatment of the anemia partially corrects this problem. There is also some evidence of impaired fibrinolysis in patients with renal disease.
In the past, the bleeding time was considered to be the most useful clinical test of coagulation in patients with renal disease, but much of the evidence supporting testing and treatment was derived from poor-quality studies performed more than 30 years ago. We now know that dialysis, especially peritoneal dialysis, improves platelet function. Erythropoietin, cryoprecipitate, conjugated estrogens, desmopressin, and tranexamic acid have all independently been shown to reduce bleeding time.38,39 In the past decade, citrate has risen in popularity as a replacement anticoagulant in continuous renal-replacement therapy, with a reduction in bleeding, although data on its safety in patients with liver failure are lacking.40
FIBRINOLYTIC BLEEDING
Excessive fibrinolysis that threatens clot integrity is known as hyperfibrinolysis.41 Abnormal fibrinolytic activity may be overlooked as a cause of bleeding, particularly in liver disease, and the condition is difficult to diagnose because of the absence of a specific routine assay. Clinical suspicion should be high in cases in which bleeding continues despite hemostatic replacement therapy, platelet levels are relatively conserved but fibrinogen levels are disproportionately low, and D-dimer levels are disproportionately high for disseminated intravascular coagulation. Thromboelastography, which may help differentiate fibrinolytic activation from coagulation factor deficiency, is a crude tool, since it detects only the most marked changes.42 Fibrinolytic bleeding should be considered particularly in patients with liver disease and disseminated cancers. The use of tranexamic acid, either by infusion or orally (depending on the severity of the problem and the state of the patient), is beneficial in controlling bleeding.
VON WILLEBRAND'S DISEASE
If unexplained bleeding occurs, consideration should be given to the late presentation of an inherited bleeding disorder. A personal and family history of easy bruising and bleeding should be sought. Occasionally, a condition such as mild von Willebrand's disease may present with persistent oozing after an injury or surgery.43
Acquired von Willebrand's disease, which can be caused by several potential mechanisms due to autoantibodies, myeloproliferative and lymphoproliferative disorders,44 or the breakdown of high-molecular-weight von Willebrand factor multimers owing to high intravascular or extracorporeal circuit shear stresses, may also occur in patients in the ICU. This disorder can also be caused by shear stresses on blood flow in extracorporeal circuits, such as those caused by extracorporeal membrane oxygenation44 and left ventricular assist devices. Intravascular shear stress from aortic-valve stenosis can cause acquired von Willebrand's disease, leading to gastrointestinal bleeding (Heyde's syndrome).45
Acquired von Willebrand's disease is treated with the use of either desmopressin, which stimulates the release of residual stores of von Willebrand factor by endothelial cells, or von Willebrand factor concentrates, with the latter considered to be the more effective therapy.46 The use of antifibrinolytic agents may be considered to alleviate mucocutaneous bleeding. Acquired von Willebrand's disease due to high shear stresses requires the removal of the cause of the condition whenever possible.
BLEEDING ASSOCIATED WITH ANTITHROMBOTIC THERAPY
TABLE 4
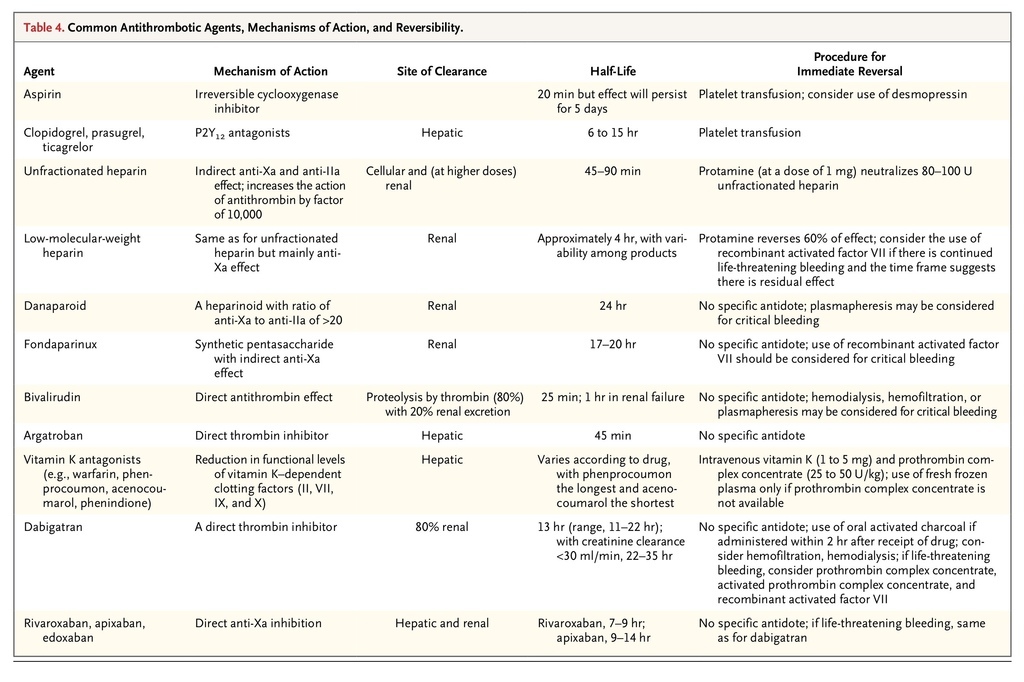
Common Antithrombotic Agents, Mechanisms of Action, and Reversibility.
summarizes the current antithrombotic drugs, their mechanisms of action, and reversibility.47,48 It is difficult to treat a bleeding patient who is receiving an oral anticoagulant such as dabigatran and rivaroxaban, since there is no specific antidote. Studies that have evaluated the reversal of the new oral anticoagulants have been limited to reversal of drug effect with the use of recombinant activated factor VII and prothrombin complex concentrate. Current evidence suggests that prothrombin complex concentrate may be the best option and that it reverses the effects of rivaroxaban better than the effects of dabigatran.49,50 General measures such as stopping the antithrombotic medication, documenting the time and amount of the last drug dose, and noting the presence of renal and hepatic impairment are suggested. Management may be aided by obtaining a full blood count and hemostatic screening, along with a specific laboratory test to measure the antithrombotic effect of the drug, if available. If the medication has been recently ingested and there is no specific antidote, oral activated charcoal may be given to absorb any residual drug in the stomach.
CONCLUSIONS
The management of bleeding in critically ill patients remains a major clinical challenge. The cause of a bleeding problem may be complex and only partially understood, with limited diagnostic tools and management strategies currently available. The absence of robust evidence from clinical trials to guide the management of acquired bleeding disorders is very striking and points to the need for studies to address the many evidence gaps that currently exist.


 留言列表
留言列表
 線上藥物查詢
線上藥物查詢 