

Volume 363:291-293
Deborah M. Muoio, Ph.D.
Metabolic disorders that are related to obesity, such as type 2 diabetes and heart disease, are intimately connected to perturbations in systemic lipid balance. Clinicians often rely on weight gain and dyslipidemia as warnings of disease risk, but lipids amassing within metabolic organs (e.g., skeletal muscle, heart, and liver) are an even stronger predictor of an adverse outcome. Indeed, several specific lipid-derived molecules that are agents of tissue dysfunction have been identified.1 Researchers who are studying lipotoxicity seek to understand the mechanisms that govern lipid balance and unravel pathways that link ectopic fat accumulation to metabolic failure.
To this end, Hagberg and colleagues2 have reported a new and unexpected lipid regulatory circuit involving crosstalk between highly oxidative tissues and their supporting vasculature. They found that vascular endothelial growth factor B (VEGFB), which is produced by heart, skeletal muscle, and brown adipose tissue, targets the local endothelium and promotes fatty acid transport from the blood, across the endothelial-cell layer, to the foregoing metabolic destinations (Figure 1). Their study establishes a novel role for VEGFB as a paracrine signal that regulates lipid delivery to fat-burning tissues, a finding that raises provocative questions about the promise of this growth factor as a therapeutic target.
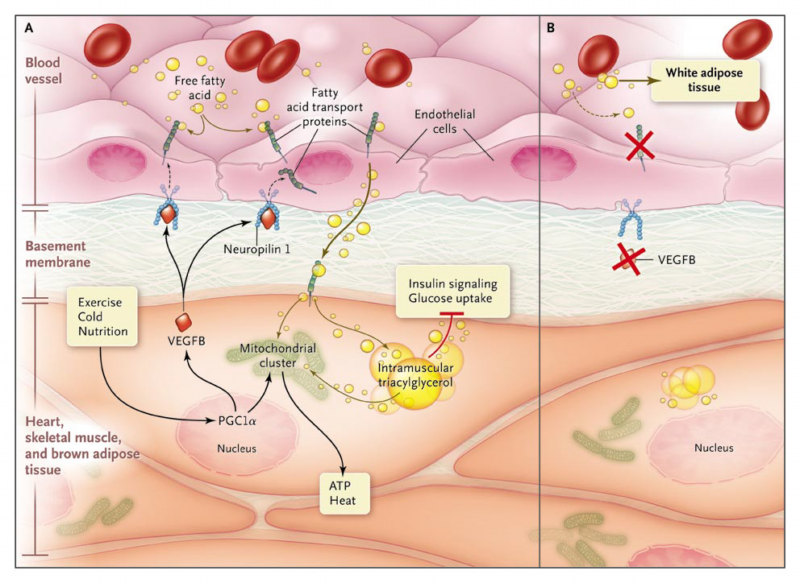
Figure 1. Fat Transport.
In Panel A, heart, skeletal muscle, and brown adipose tissue burn fatty acids to support contraction and heat production. Physiological stimuli (exercise, cold exposure, and nutrition) activate master transcriptional regulators, such as peroxisome proliferator-activated receptor coactivator 1 (PGC1), that induce mitochondrial genesis and the production of vascular endothelial growth factor B (VEGFB). VEGFB acts on neighboring endothelial cell–surface receptors, such as neuropilin 1, to stimulate the expression of fatty acid transport proteins (FATP), which facilitates the uptake of fatty acids from the blood across the endothelial-cell layer. This paracrine circuit thereby coordinates oxidative metabolism with vascular delivery of the lipid substrate. Targeting of this pathway with a drug might hold promise for the treatment of diabetes mellitus, because inappropriate accumulation of intramuscular triacylglycerol and other lipid species impairs insulin signaling and glucose uptake. In Panel B, genetic ablation of VEGFB enhances glucose disposal in muscle but at the expense of increased adiposity, because the extra fatty acids are redirected to white adipose tissue.
Although VEGFB belongs to a family of secreted vasculogenic regulators, unlike its close relatives VEGFA and placental growth factor, it has surprisingly weak angiogenic potency.3 Hagberg et al. gained a clue to its biologic function on interrogating publicly available microarray data on gene-expression patterns. They observed that the mouse orthologue of the gene encoding VEGFB, Vegfb, clustered with a large network of nuclear-encoded mitochondrial genes, suggesting that Vegfb and the mitochondrial genes are coregulated. Because mice that are completely deficient in Vegfb have normal mitochondrial mass,2 the authors attributed the strong correlation between the expression of Vegfb and that of the mitochondrial cluster to a common transcriptional switch. Unpublished data pointed toward estrogen-related receptor and its coactivator, peroxisome proliferator-activated receptor coactivator 1 (PGC1), which are both prominent master regulators of mitochondrial programming, as candidates. Hagberg et al. established a unique role for VEGFB in fatty acid metabolism by showing that the growth factor is expressed most abundantly in mitochondria-enriched tissues (e.g., heart, skeletal muscle, and brown adipose tissue); that treatment of cultured endothelial cells with VEGFB or forced expression of the protein in mouse heart tissue stimulated vascular expression of several distinct fatty acid transport proteins, concomitant with elevated transendothelial lipid transport; that Vegfb knockout mice had decreased uptake and storage of fatty acid in heart, skeletal muscle, and brown adipose tissue; and that the metabolic actions of VEGFB require signaling through VEGF receptor 1 and neuropilin 1, two cell-surface receptors that are abundantly expressed in endothelial cells.
This study has several implications. First, the results add to indisputable evidence that lipid balance at the whole-body and tissue levels is profoundly reliant on interorgan crosstalk. Myocytes and brown adipocytes are heavy consumers of fatty acids. Abundant expression of VEGFB enables these cells to relay this substrate preference to the local vessels that deliver their nutrient supply. As such, VEGFB is likely to facilitate normal physiological shifts in metabolic currency. For example, physical activity and cold exposure increase fat catabolism by muscle and brown adipose tissue, respectively. Repeated exposure to these stimuli activates PGC1, which triggers mitochondrial genesis and reinforces the resident oxidative machinery.4 Exercise-trained muscles also adapt by boosting triacylglycerol storage, ensuring a readily available substrate pool to support the high costs of contractile activity. Thus, whereas VEGFA is known to mediate contraction-induced angiogenesis,5 VEGFB might serve a complementary role by instructing the expanding capillary bed to shuttle lipid fuels toward working muscles.
In contrast to a healthy condition in which the delivery of fatty acids is appropriately matched to metabolic needs, disease states manifest ectopic lipid accumulation that grossly exceeds energy demand. Is VEGFB a contributor in such instances? Support for this possibility comes from the observation that mice consuming a long-term high-fat diet had elevated levels of Vegfb messenger RNA in heart tissue.2 Thus, obesity and its attendant metabolic abnormalities might induce maladaptive expression of VEGFB that in turn exacerbates lipotoxicity and consequent tissue dysfunction.
Would blockade of VEGFB have therapeutic value? As predicted, genetic ablation of VEGFB lowered intramuscular content of neutral lipids, a presumably positive outcome, given that surplus fat impairs insulin signaling and glucose tolerance in sedentary muscles.1 Accordingly, Vegfb-deficient mice had increased glucose uptake into heart tissue, a finding suggestive of antidiabetic potential. On the other hand, Vegfb knockout mice were fatter because lipids that were turned away by muscle and brown adipose tissue were rerouted to white adipose tissue. What does this mean from a clinical viewpoint? Although a clear picture of the net metabolic effect of inactivating VEGFB awaits further study, the results imply that without a change in caloric intake or expenditure, drugs that are designed to dissuade fat uptake into one tissue are likely to redistribute the energetic burden to another site. Perhaps the simplest interpretation at this stage is that the pharmacotherapeutic ideal of "having our cake and eating it too" mistakenly dismisses the first law of thermodynamics. Instead, the clinically proven, albeit less enticing, approach to combating lipotoxicity is the one that calls for less cake and more activity.
References
Muoio DM, Newgard CB. Mechanisms of disease: molecular and metabolic mechanisms of insulin resistance and beta-cell failure in type 2 diabetes. Nat Rev Mol Cell Biol 2008;9:193-205. [CrossRef][Web of Science][Medline]
Hagberg CE, Falkevall A, Wang X, et al. Vascular endothelial growth factor B controls endothelial fatty acid uptake. Nature 2010;464:917-921. [CrossRef][Web of Science][Medline]
Breen EC. VEGF in biological control. J Cell Biochem 2007;102:1358-1367. [CrossRef][Web of Science][Medline]
Handschin C, Spiegelman BM. Peroxisome proliferator-activated receptor gamma coactivator 1 coactivators, energy homeostasis, and metabolism. Endocr Rev 2006;27:728-735. [Free Full Text]
Olfert IM, Howlett RA, Tang K, et al. Muscle-specific VEGF deficiency greatly reduces exercise endurance in mice. J Physiol 2009;587:1755-1767. [Free Full Text]


 留言列表
留言列表
 線上藥物查詢
線上藥物查詢 