


Obstructive sleep apnea is a common disorder, characterized by recurrent narrowing and closure of the upper airway accompanied by intermittent oxyhemoglobin desaturation and sympathetic activation.1 Sequelae include excessive sleepiness and impaired quality of life. Moderate-to-severe obstructive sleep apnea, defined as an apnea–hypopnea index (AHI) score of 15 or more apnea or hypopnea events per hour, is an independent risk factor for insulin resistance, dyslipidemia, vascular disease, and death.2-7 Treatment with continuous positive airway pressure (CPAP) with the use of a mask favorably modifies these adverse health consequences.8 However, the general effectiveness of CPAP therapy is dependent on patient acceptance of and adherence to the treatment.9,10
Alternative treatments to CPAP include custom-made oral-appliance therapy and a variety of upper-airway surgeries.11,12 Since evidence-based reviews do not uniformly support the efficacy of these treatments for moderate-to-severe sleep apnea, new therapy is desirable.13,14
The onset of apnea is accompanied by a reduction in drive to the upper-airway muscles,15,16 and upper-airway patency is strongly correlated with the activation of the genioglossus muscle.17 Upper-airway stimulation with the use of unilateral stimulation of the hypoglossal nerve has been developed as a possible treatment option and has shown promise in feasibility trials.18-23
Using a multicenter, prospective, single-group trial design followed by a randomized, therapy-withdrawal trial that included only participants who had had a response to therapy, we addressed the clinical safety and effectiveness of upper-airway stimulation at 12 months after implantation. This technology permits stimulation to be synchronized with ventilatory effort during sleep.
METHODS
Participants
Participants with moderate-to-severe obstructive sleep apnea were eligible for enrollment if they had difficulty accepting or adhering to CPAP treatment. Exclusion criteria were a body-mass index (BMI; the weight in kilograms divided by the square of the height in meters) of more than 32.0, neuromuscular disease, hypoglossal-nerve palsy, severe restrictive or obstructive pulmonary disease, moderate-to-severe pulmonary arterial hypertension, severe valvular heart disease, New York Heart Association class III or IV heart failure, recent myocardial infarction or severe cardiac arrhythmias (within the past 6 months), persistent uncontrolled hypertension despite medication use, active psychiatric disease, and coexisting nonrespiratory sleep disorders that would confound functional sleep assessment.
Study Design and Oversight
The study was designed by the sponsor (Inspire Medical Systems), the investigators, and the Food and Drug Administration as a multicenter, prospective, single-group trial with participants serving as their own controls. There was no concurrent control group. The primary outcome evaluation was followed by a randomized, controlled therapy-withdrawal study that included a subgroup of consecutive participants selected from the population that had a response to therapy.
The trial protocol was approved by the institutional review board (in the United States) or medical ethics committee (in Europe) at each participating center. All the participants provided written informed consent before enrollment. An independent clinical-events committee and a data and safety monitoring board provided review and adjudication of safety data. Verification of source data was performed by independent monitors. The study investigators had full access to the data and had the right to submit the manuscript for publication without input from the sponsor. The writing committee (the first, second, and last authors), an independent statistician (Teri Yurik, NAMSA), and the sponsor vouch for the accuracy and completeness of the data and analyses and for the fidelity of the study to the protocol.
The primary outcome measures were assessed by means of overnight polysomnography and scored by an independent core laboratory with the use of standard criteria.24 The data analysis was performed by the independent statistician, with the results reviewed by the first and last authors. The first author wrote the manuscript with assistance from the writing committee; no one who is not listed as an author contributed substantially to the study report.
Screening and Implantation
In order for investigators to verify eligibility for the implantation, enrolled participants underwent screening that included polysomnography, medical and surgical consultation, and endoscopy during drug-induced sleep.25 Participants were excluded if the AHI score from the screening polysomnography was less than 20 or more than 50 events per hour, if central or mixed sleep-disordered breathing events accounted for more than 25% of all apnea and hypopnea episodes, or if the AHI score while the person was not in a supine position was less than 10 events per hour. Participants were also excluded if pronounced anatomical abnormalities preventing the effective use or assessment of upper-airway stimulation were identified during the surgical consultation (e.g., tonsil size of 3 or 4 [tonsils visible beyond the pillars or extending to midline]) or if complete concentric collapse at the retropalatal airway was observed on endoscopy performed during drug-induced sleep.25
Qualified participants underwent a surgical procedure to implant the upper-airway stimulation system (Inspire Medical Systems)20 (Figure 1
FIGURE 1
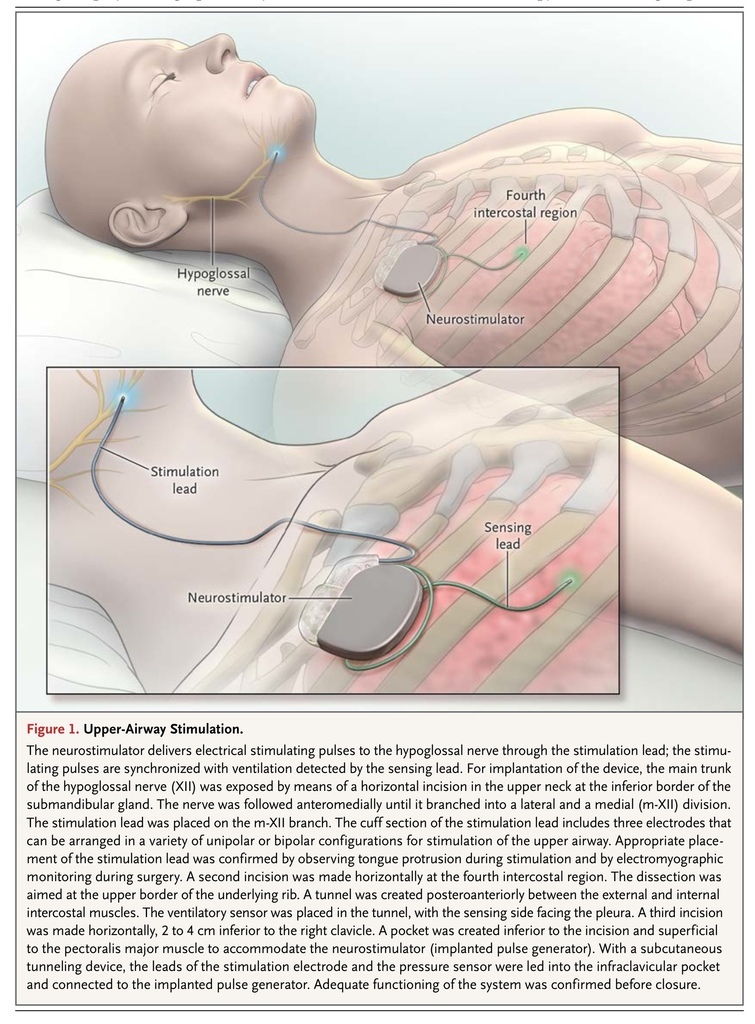
Upper-Airway Stimulation.
). The stimulation electrode was placed on the hypoglossal nerve to recruit tongue-protrusion function; the sensing lead was placed between the internal and external intercostal muscles to detect ventilatory effort; the neurostimulator was implanted in the right ipsilateral mid-infraclavicular region.
Approximately 1 month after implantation, all the participants underwent a second baseline diagnostic polysomnographic examination before activation of the device. Immediately after this polysomnography, all the participants had their device activated and were instructed regarding the use of a controller to initiate and terminate therapy on a nightly basis. After activation, participants had scheduled outpatient visits at months 2, 3, 6, 9, and 12; at each of these visits data on adverse events were obtained and device interrogation was performed.
Outcome Measures
The primary outcome was the change in the severity of obstructive sleep apnea in the study population, as assessed by means of the AHI and the oxygen desaturation index (ODI; the number of times per hour of sleep that the blood oxygen level drops by ≥4 percentage points from baseline). The coprimary outcome was the proportion of participants with a response from baseline to 12 months with respect to the primary outcome measures of the AHI and ODI scores. A response as measured by means of the AHI was defined as a reduction of at least 50% from baseline in the AHI score and an AHI score on the 12-month polysomnography of less than 20 events per hour.26 The ODI was chosen as a stable integrative outcome value of all forms of sleep-disordered breathing. A response as measured by means of the ODI was defined as a reduction of at least 25% from baseline in the ODI score. The prespecified primary efficacy objectives were response rates of at least 50%, as assessed by means of the AHI and ODI. All participants who received an implant were included in the primary outcome analysis; participants who did not complete the 12-month visit were considered not to have had a response.
Secondary outcome measures included self-reported sleepiness and disease-specific quality of life as assessed with the use of the Epworth Sleepiness Scale (scores range from 0.0 to 24.0, with higher scores indicating more daytime sleepiness), disease-specific quality of life, as assessed with the use of the Functional Outcomes of Sleep Questionnaire (FOSQ; scores range from 5.0 to 20.0, with higher scores indicating greater functioning), and the percentage of sleep time with the oxygen saturation less than 90%.
Follow-up
The follow-up visits at months 2, 6, and 12 included a polysomnographic study and evaluation of daytime sleepiness by means of the Epworth Sleepiness Scale. An Epworth Sleepiness Scale score of less than 10.0 is considered to be the threshold for normal subjective sleepiness.27 Participants also completed the FOSQ, on which a score of more than 17.9 is considered to be the threshold for persons with normal sleep-related quality of life. A change of 2.0 or more points in the FOSQ score is considered to indicate a clinically meaningful improvement in daily functioning.28
During the polysomnographic studies at 2 months and 6 months, device variables were adjusted with the use of a programmer unit that communicates with the device by means of telemetry. The adjusted variables included the stimulation voltage, rate, and pulse width and the timing of electrical stimulation. No device adjustments were made in the 30 days before or during the polysomnographic study at 12 months.
At the 12-month visit, the first 46 consecutive participants who met the criterion of having a response to therapy were randomly assigned, in a 1:1 ratio, to the therapy-maintenance group or the therapy-withdrawal group.29 This design filtered out persons who had not had a response to therapy. The therapy-withdrawal group had the device turned off for 7 days, whereas the therapy-maintenance group continued with the device turned on. Polysomnography was performed after the randomization period to measure the effects of therapy withdrawal, as compared with continued use of the therapy. For the 12-month nonrandomized phase of the study, participant enrollment commenced on November 10, 2010, and ended on March 23, 2013.
Adverse Events
Adverse events were reported and then reviewed and coded by the clinical-events committee. Serious adverse events were defined as any events that led to death, life-threatening illness, permanent impairment, or new or prolonged hospitalization with serious health impairment.
Statistical Analysis
For the coprimary outcomes, the AHI and ODI scores at the 12-month follow-up were compared with the baseline measurements, which were the averages of the measurements obtained before implantation and at the 1-month preactivation visit, to determine a binary outcome of status with respect to response to the therapy. We estimated that 108 participants would need to be enrolled for the study to have 80% power to evaluate the primary outcome, with the exact one-sided binomial test set at a significance level of 2.5%. The changes in the Epworth Sleepiness Scale and FOSQ scores from the preimplantation screening to the 12-month visit were calculated for each participant. P values from a paired t-test were calculated for the secondary outcome measures.
In the randomized controlled therapy-withdrawal trial, the difference in mean AHI scores (i.e., the difference between scores obtained at the 12-month visit in the nonrandomized phase and those obtained at the end of the randomized phase) was compared between the therapy-maintenance group and the therapy-withdrawal group. We estimated that 40 participants would need to undergo randomization in a 1:1 ratio in order for the study to have 80% power to detect a significant difference between groups, at the 5% significance level, with the use of a two-sided t-test.
RESULTS
Characteristics of the Participants
The study population consisted of 126 participants (83% of whom were men), with a mean age of 54.5 years (range, 31 to 80) and mean BMI of 28.4 (range, 18.4 to 32.5). Per protocol, all the participants had a history of nonadherence to CPAP therapy; 17% of the participants had undergone a uvulopalatopharyngoplasty (surgery to remove excess upper-airway tissue) for the treatment of obstructive sleep apnea.
The mean time from the diagnosis of obstructive sleep apnea to study enrollment was 5.6 years. The mean AHI score on preimplantation screening polysomnography was 32.0 events per hour, and the mean ODI score was 28.9 events per hour. At the baseline visit before implantation, the mean FOSQ score was 14.3, and the mean Epworth Sleepiness Scale score was 11.6. The mean AHI score on the second baseline polysomnography was 31.9 events per hour. There was no significant difference between the two baseline AHI assessments (P=0.83).
A total of 124 of 126 participants (98%) completed the follow-up at 12 months. The mean BMI at 12 months was 28.5, which did not differ significantly from the mean BMI at baseline. The characteristics of the study cohort at baseline are presented in Table 1
TABLE 1
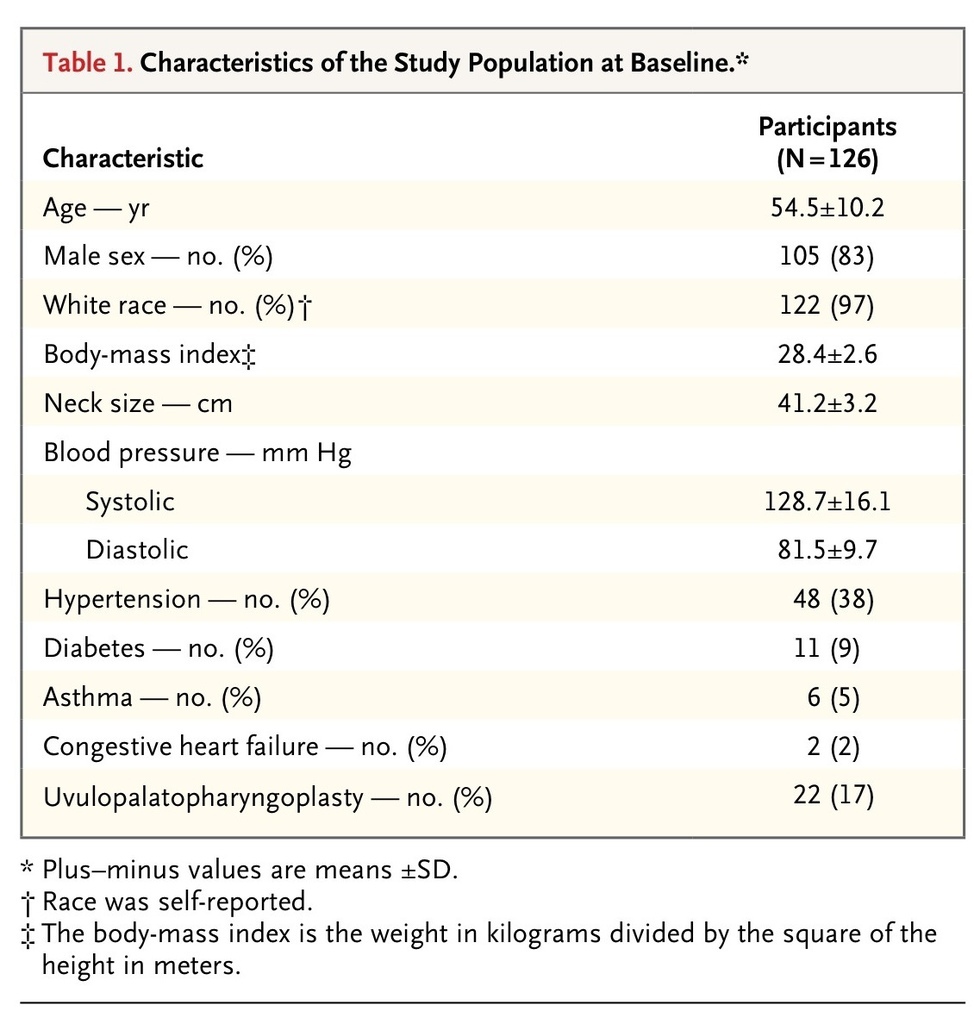
Characteristics of the Study Population at Baseline.
. Information on study enrollment, randomization, and follow-up are shown in Figure 2
FIGURE 2
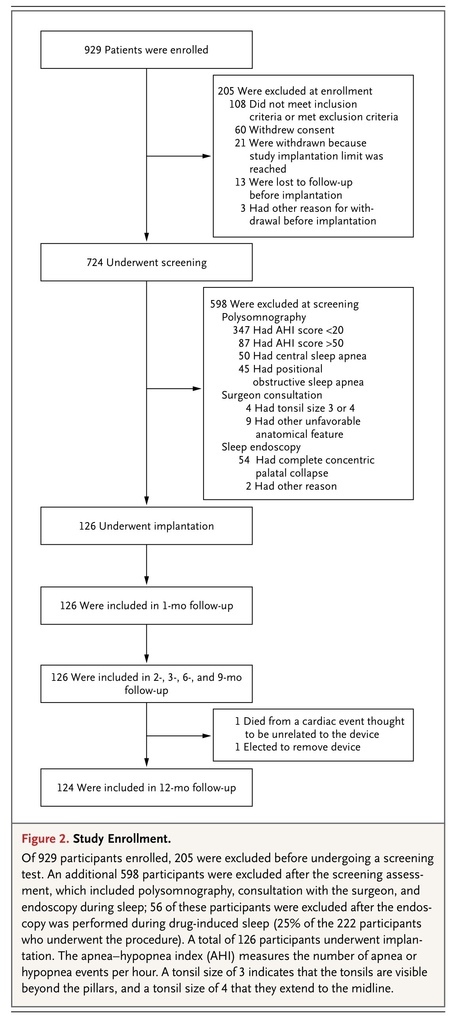
Study Enrollment.
.
Surgical Implantation
The upper-airway stimulation device was successfully implanted in all 126 participants. The median time for surgical implantation was 140 minutes (range, 65 to 360). Participants were discharged after surgery on the same day (16% of participants), the next day (79%), or the second day after surgery (5%).
Primary Outcomes
The scores on the AHI and ODI (primary outcome measures) were lower (indicating fewer episodes of sleep apnea) at 12 months than at baseline. The median AHI score decreased 68%, from the baseline value of 29.3 events per hour to 9.0 events per hour. The median ODI score decreased 70%, from 25.4 events per hour to 7.4 events per hour. At the 12-month visit, the criteria for the coprimary outcome of a reduction of at least 50% in the AHI score from baseline and an AHI score of less than 20 events per hour were met by 66% of the participants (83 of 126 participants; lower boundary of the 97.5% confidence interval [CI], 57). The criterion for the coprimary outcome of a reduction of at least 25% in the ODI score from baseline was met by 75% of participants (94 of 126; lower boundary of the 97.5% CI, 66). Both primary efficacy outcomes exceeded the predefined study objectives (Table 2
TABLE 2
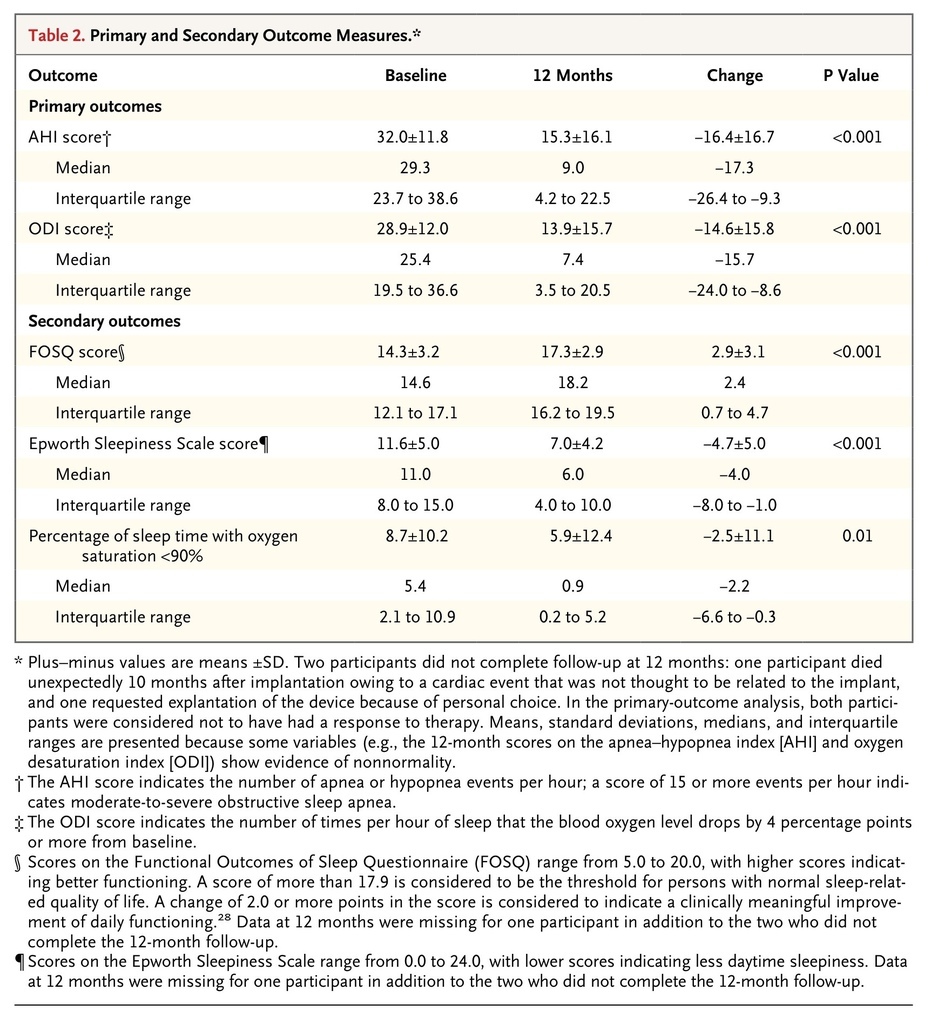
Primary and Secondary Outcome Measures.
).
Secondary Outcomes
Scores on the FOSQ and Epworth Sleepiness Scale indicated significant improvement at 12 months, as compared with baseline. The increase in the FOSQ score (mean change, 2.9 points; 95% CI, 2.4 to 3.5) exceeded the 2.0-point increase that is typically considered to be a clinically meaningful improvement. Similarly, the Epworth Sleepiness Scale score at 12 months was consistent with normalization of the measure (i.e., score <10.0). The median percentage of sleep time with the oxygen saturation less than 90% decreased from a baseline value of 5.4% to 0.9% at 12 months (Table 2).
Therapy-Withdrawal Study
Among the 46 consecutive participants with a response to therapy who underwent randomization, the demographic and clinical characteristics at baseline were similar with regard to age, BMI, neck size, and AHI and ODI scores. By design, participants who had not had a response were not included in this part of the study.
The AHI and ODI scores were similar in the two groups at 12 months (baseline of the randomized portion of the trial). There was a significant difference between the therapy-withdrawal group and the therapy-maintenance group with respect to the change in AHI scores from the beginning of the randomization period at 12 months to the assessment 1 week later. Among the 23 participants in the therapy-withdrawal group, the AHI score was significantly higher at the 1-week assessment than it was at the start of the randomized phase (25.8 vs. 7.6 events per hour, P<0.001). The average increase in the AHI score in the therapy-withdrawal group was 18.2 events per hour, whereas the average increase in the therapy-maintenance group was 1.7 events per hour (difference in changes in mean scores, 16.4±12.0 events per hour; P<0.001). A similar effect was observed with respect to the mean ODI scores (Figure 3
FIGURE 3
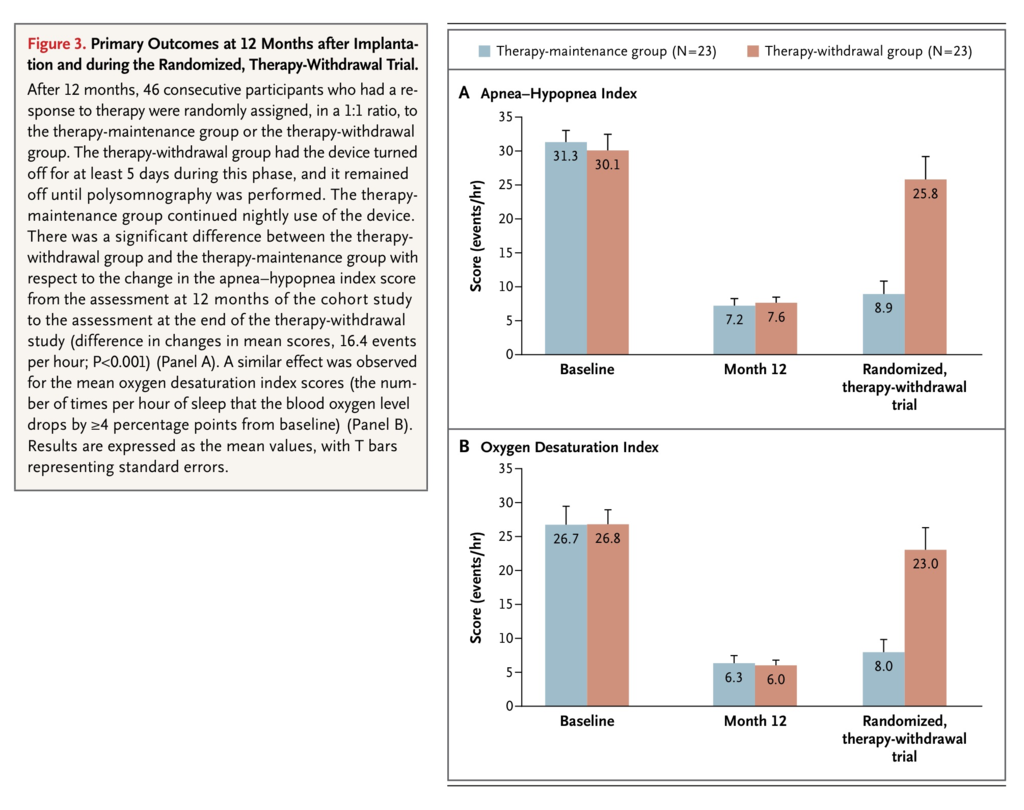
Primary Outcomes at 12 Months after Implantation and during the Randomized, Therapy-Withdrawal Trial.
).
Adverse Events
Two participants had a serious device-related adverse event requiring repositioning and fixation of the neurostimulator to resolve discomfort. A total of 33 serious adverse events not considered to be related to the implantation procedure or implanted devices were reported. Most of nonserious adverse events related to the procedure (88%) occurred within 30 days after implantation and were expected postsurgical events, including sore throat from intubation, pain at the incision site, and muscle soreness.
A total of 18% of the participants had temporary tongue weakness after surgery, which resolved over a period of days to weeks. No permanent tongue weakness was reported during the study. Among device-related events that were not considered to be serious, 40% of the participants reported some discomfort associated with stimulation, and 21% reported tongue soreness, including abrasion on the lower side of the tongue. These events were related to the functional stimulation of the tongue muscles and the resulting tongue motion over the lower teeth. Most of these events resolved after the participants acclimated to the upper-airway stimulation therapy or after the device was reprogrammed to adjust the stimulation variables. In nine participants, a tooth guard was used to resolve tongue soreness or abrasion related to the device.
The overall rate of serious adverse events was less than 2%. A detailed list of adverse events is provided in Table S1 in the Supplementary Appendix, available with the full text of this article at NEJM.org.
DISCUSSION
Patients with moderate-to-severe obstructive sleep apnea may not have consistent clinical benefit from CPAP therapy owing to poor adherence to treatment.30 These patients, if left untreated, remain at considerable risk for cardiovascular complications and death. In the current study, unilateral stimulation of the hypoglossal nerve, synchronous with ventilation, resulted in significant and clinically meaningful reductions in the severity of obstructive sleep apnea and self-reported sleepiness and improvements in quality-of-life measures at 1 year. The observed response rates, which were based on the primary outcome measures of AHI and ODI, consistently exceeded the previously defined threshold for surgical success.12 The reduction in sleepiness and improvement in quality-of-life measures at 12 months were similar to previously reported effects of CPAP on moderate-to-severe obstructive sleep apnea.28
The effect of stimulation of the hypoglossal nerve with respect to obstructive events was first described by Schwartz et al. in 1993 in a feline model.31 Subsequent studies showed that stimulation of the genioglossal muscle or the hypoglossal nerve could reverse inspiratory flow limitation during sleep.17 The current study extended the observations that were reported by Eastwood et al. over a period of 6 months in a single-group interventional trial.18 The feasibility studies conducted by our team identified a BMI of 32 or lower or an AHI score of 50 events per hour or less as phenotypic risk factors that favorably affect the success of upper-airway stimulation.22,25 This approach may not be appropriate for persons with excessive airway collapsibility.32 Screening potential participants by means of endoscopy during drug-induced sleep helped to identify functional upper-airway collapse that was likely to be focused on the retrolingual region and therefore amenable to forward motion of the base of the tongue by means of neurostimulation.25
Surgical implantation of the upper-airway stimulation system was performed by otolaryngologists at 22 academic and private centers. None of the implantation procedures resulted in serious complications, participant rehospitalizations, or explantations because of infection. The serious adverse events in the two participants who required repositioning and fixation of the neurostimulator occurred 30 days after implantation and were related primarily to discomfort at the device location. The electrical stimulation of the hypoglossal nerve evokes a functional response of the tongue muscles and an anterior displacement of the tongue. The patient can feel the anterior displacement of the tongue during wakefulness when the stimulation is turned on. Similar to CPAP, therapeutic stimulation variables were determined during attended in-laboratory sleep studies.
The implanted upper-airway stimulation device eliminated adherence issues associated with wearing a CPAP mask. The daily use of upper-airway stimulation was 86%, as assessed on the basis of self-report (see the Supplementary Appendix). Objective use of the device, quantified as the time spent using the device each night, could not be directly reported with the current generation of the device. The average stimulation time per night was measured. This value accounts for the time predominately associated with the inspiratory phase of the breathing cycle. Assuming a normal duty cycle of 1:2.0 or 1:1.5, the average objective use would be in excess of 5 hours per night (see the Supplementary Appendix). Additional objective data on adherence will be required to confirm the findings of the current study.
The current study was designed to assess the severity and symptoms of obstructive sleep apnea before the implantation of the upper-airway stimulation device as compared with 12 months after implantation, with the use of a prospective single-group trial design in which the participants served as their own controls. Only participants who could not use CPAP, or who declined to do so, were recruited for the study. A control group of therapeutic CPAP users (i.e., a comparative-effectiveness design) would be impractical, given the current study design.
Some participants had a significant increase in the AHI score at month 12 (see the Supplementary Appendix). An additional analysis of the association between the baseline characteristics and outcome measures did not identify predictors that differentiated between participants who had a response and those who did not.
The randomized, controlled therapy-withdrawal study in which some participants had the therapy turned off for 1 week provided evidence that the therapeutic effect established at 12 months was attributable to the upper-airway stimulation therapy, rather than variability in the AHI score. The randomized phase included only consecutive participants who had had a response to therapy and, as a result, does not provide information on participants who did not have a response to therapy.
By design, this trial enrolled participants with moderate-to-severe obstructive sleep apnea who had various difficulties adhering to CPAP and who did not have clinically significant central or mixed sleep apnea or complete concentric collapse at the retropalatal airway on endoscopy during drug-induced sleep. The cohort had a reduction in the severity of obstructive sleep apnea, and the adverse-event profile was acceptable.


 留言列表
留言列表
 線上藥物查詢
線上藥物查詢 