
Astaxanthin有效抑制脈絡膜新血管增生
Investigative Ophthalmology & Visual Science April 2008, Vol.49, 1679-1685. doi:10.1167/iovs.07-1426
Retinal Cell Biology | April 2008
|
Abstract
purpose. Astaxanthin (AST) is a carotenoid found in marine animals and vegetables. The purpose of the present study was to investigate the effect of AST on the development of experimental choroidal neovascularization (CNV) with underlying cellular and molecular mechanisms.
methods. Laser photocoagulation was used to induce CNV in C57BL/6J mice. Mice were pretreated with intraperitoneal injections of AST daily for 3 days before photocoagulation, and treatments were continued daily until the end of the study. CNV response was analyzed by volumetric measurements 1 week after laser injury. Retinal pigment epithelium–choroid levels of IκB-α, intercellular adhesion molecule (ICAM)-1, monocyte chemotactic protein (MCP)-1, interleukin (IL)-6, vascular endothelial growth factor (VEGF), VEGF receptor (VEGFR)-1, and VEGFR-2 were examined by Western blotting or ELISA. AST was applied to capillary endothelial (b-End3) cells, macrophages, and RPE cells to analyze the activation of NF-κB and the expression of inflammatory molecules.
results. The index of CNV volume was significantly suppressed by treatment with AST compared with that in vehicle-treated animals. AST treatment led to significant inhibition of macrophage infiltration into CNV and of the in vivo and in vitro expression of inflammation-related molecules, including VEGF, IL-6, ICAM-1, MCP-1, VEGFR-1, and VEGFR-2. Importantly, AST suppressed the activation of the NF-κB pathway, including IκB-α degradation and p65 nuclear translocation.
conclusions. AST treatment, together with inflammatory processes including NF-κB activation, subsequent upregulation of inflammatory molecules, and macrophage infiltration, led to significant suppression of CNV development. The present study suggests the possibility of AST supplementation as a therapeutic strategy to suppress CNV associated with AMD.
|
Age-related macular degeneration (AMD) is the most common cause of blindness in developed countries.
1 It is complicated by choroidal neovascularization (CNV), leading to severe vision loss and blindness.
2 Molecular and cellular mechanisms underlying CNV are not fully elucidated. CNV seen in AMD develops with oxidative stress and chronic inflammation adjacent to RPE, Bruch membrane, and choriocapillaris.
3 4 Recent experimental and clinical studies have indicated vascular endothelial growth factor (VEGF) as critical for promoting CNV.
5 6 CNV formation is associated with the influx of inflammatory cells including macrophages, which are the rich source of VEGF. Pharmacologic depletion of macrophages, present in human and murine CNV tissues,
2 7 8 9resulted in significant suppression of murine CNV.
7 9 CNV tissues from human surgical samples and the rodent laser-induced model express inflammation-related molecules, including intercellular adhesion molecule (ICAM)-1.
10 11 Genetic ablation of ICAM-1 or CC chemokine receptor-2, a receptor for monocyte chemotactic protein (MCP)-1, inhibited CNV in the murine model.
8 10 We have also highlighted the inflammatory mechanisms mediated by the rennin–angiotensin system
12 13 and interleukin (IL)-6 receptor signaling,
14demonstrating the regulation of inflammation as an important therapeutic strategy to suppress CNV.
Carotenoids are a family of more than 700 natural lipid-soluble pigments produced only by phytoplankton, algae, plants, and a limited number of fungi and bacteria. Astaxanthin (AST) is one of the most prevalent carotenoids and is abundantly present in the red pigment of crustacean shells (e.g., crabs, shrimp), salmon, and asteroidean.
15 Several previous studies have demonstrated that AST exhibits a wide variety of biological activities, including antioxidative,
16 anti-
Helicobacter pylori,
17 anticancer,
18 and anti-inflammatory effects.
19 2021 As an anti-inflammatory agent, AST has been shown to inhibit lipopolysaccharide (LPS)-induced ocular
19 20 and systemic
21 inflammation. No data have been reported, however, that show the effects of AST on RPE–choroid inflammation and subsequent CNV generation. Here we report the first evidence of the antipathogenic role of AST on CNV development, together with underlying molecular and cellular mechanisms.
Methods
Animals
Male C57BL/6J mice (CLEA, Tokyo, Japan) 6 to 8 weeks of age were used. All animal experiments were conducted in accordance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research.
Induction of CNV
Laser-induced CNV is widely used as an animal model for wet AMD and reflects the pathogenesis of inflammation-related CNV seen in AMD. In this model, new vessels from the choroid invade the subretinal space after photocoagulation. Laser photocoagulation was performed at five spots per eye around the optic disc (wavelength, 532 nm; power, 200 mW; duration, 100 ms; spot size, 75 μm) using a slit lamp delivery system (NOVUS Spectra; Lumenis, Tokyo, Japan), as described previously.
7
Treatment with AST
Animals were pretreated with AST (Sigma, St. Louis, MO) or phosphate-buffered saline (PBS) containing 0.1% dimethyl sulfoxide (DMSO) as vehicle daily for 3 days before photocoagulation, and the treatments were continued daily until the end of the study. AST was intraperitoneally administered to mice at the dose of 1, 10, or 100 mg/kg body weight (BW).
Quantification of Laser-Induced CNV
One week after laser injury, eyes were enucleated and fixed with 4% paraformaldehyde (PFA). Eyecups obtained by removing anterior segments were incubated with 0.5% fluorescein-isothiocyanate (FITC)-isolectin B4 (Vector, Burlingame, CA). CNV was visualized with blue argon laser wavelength (488 nm) using a scanning laser confocal microscope (FV1000; Olympus, Tokyo, Japan). Horizontal optical sections of CNV were obtained at every 1-μm step, from the surface to the deepest focal plane. The area of CNV-related fluorescence was measured by National Institutes of Health Image software. Summation of the whole fluorescence area was used as the index of CNV volume, as described previously.
7
Immunohistochemistry for Infiltrating Macrophages
Wholemount choroid–sclera complexes obtained 3 days after photocoagulation were incubated with a goat polyclonal antibody against mouse PECAM-1 (CD31; Santa Cruz Biotechnology, Santa Cruz, CA) and a rat polyclonal antibody against F4/80 (AbD Serotec, Raleigh, NC). Avidin-fluorescent dye (Alexa 488 and Alexa 546; Invitrogen, Carlsbad, CA)–tagged secondary antibodies were then applied, as described previously.
14
Quantification of Infiltrating Macrophages
Total RNA was isolated from the RPE–choroid complex 3 days after photocoagulation and reverse transcribed. Quantitative polymerase chain reaction (PCR) analyses for F4/80 and glyceraldehydes-3-phosphate dehydrogenase (GAPDH) were performed in a real-time PCR system (ABI 7500 Fast; Applied Biosystems, Foster, CA) in combination with TaqMan chemistry, as described previously.
22 23 Relative expression was then calculated as the density of the product of F4/80 divided by that for GAPDH from the same cDNA.
Enzyme-Linked Immunosorbent Assay
The RPE–choroid complex was carefully isolated from the eyes 3 days after photocoagulation and placed into 100 μL lysis buffer (0.02 M HEPES, 10% glycerol, 10 mM Na4P2O7, 100 μM Na3VO4, 1% Triton, 100 mM NaF, 4 mM EDTA [pH 8.0]) supplemented with protease inhibitors (2 mg/L aprotinin, 100 μM phenylmethylsulfonyl fluoride, 10 μM leupeptin, 2.5 μM pepstatin A) and sonicated. The lysate was centrifuged at 15,000 rpm for 15 minutes at 4°C, and the levels of ICAM-1, MCP-1, IL-6, VEGF, VEGF receptor (VEGFR)-1 and VEGFR-2 were determined with the ICAM-1, MCP-1, IL-6, VEGF, VEGFR-1, and VEGFR-2 ELISA kits (R&D Systems, Minneapolis, MN) according to the manufacturer’s protocols.
Western Blot Analyses for IκB-alpha
Protein extracts were obtained from the homogenized RPE–choroid complex 4 hours after photocoagulation. Each sample containing 30 μg total protein was separated by SDS-PAGE and electroblotted to polyvinylidene fluoride (PVDF) membrane (ATTO, Tokyo, Japan). After blocking nonspecific binding with 5% skim milk, the membranes were incubated with a rabbit monoclonal antibody against IκB-α (Cell Signaling Technology, Beverly, MA) or an anti-α-tubulin antibody (1:2000; Sigma) at 4°C overnight. Membranes were then incubated with biotin-conjugated secondary antibodies followed by avidin-biotin complex (1:2000; Vectastain ABC Elite Kit; Vector Laboratories, Burlingame, CA) or a horseradish peroxidase-conjugated goat antibody against rabbit IgG (1:5000; BioSource, Camarillo, CA). The signals were visualized with an ECL kit (GE Healthcare, Buckinghamshire, UK) according to the manufacturer’s protocol.
In Vitro Assays
We examined the in vitro effect of AST on inflammatory responses from three major cell types associated with CNV formation—microvascular endothelial cells, macrophages and RPE cells—using the murine cell lines b-End3 and RAW264.7 and the human cell line ARPE-19, respectively. Cells were pretreated with AST (50 or 150 μM) or vehicle in serum-starved DMEM for b-End3 and RAW264.7 cells or DMEM/F12 (Sigma) for ARPE-19 cells. Pretreated cells were then stimulated with tumor necrosis factor (TNF)-α (10 ng/mL for b-End3 cells and 20 ng/mL for ARPE-19 cells; Sigma) or LPS (200 ng/mL for RAW264.7 cells; Sigma) plus AST (50 or 150 μM) or vehicle. After 30-minute incubation, the cell lysates were processed for Western blot analyses for IκB-α. After 6-hour incubation, the cell lysate from b-End3 and RAW264.7 cells were processed for ELISA for ICAM-1, VEGFR-2, and VEGFR-1. Supernatants from b-End3 and RAW264.7 cells were processed for ELISA for MCP-1, IL-6, and VEGF. After 24-hour incubation, the supernatant from ARPE-19 cells was processed for ELISA for MCP-1 and VEGF.
Immunocytochemistry for NF-κB p65
After 24-hour pretreatment with AST (150 μM) or vehicle in serum-starved DMEM for b-End3 cells and RAW264.7 cells or DMEM/F12 for ARPE-19 cells, cells were incubated with TNF-α (10 ng/mL for b-End3 cells and 20 ng/mL for ARPE-19 cells) or LPS (200 ng/mL for RAW264.7 cells; Sigma) with AST (150 μM) or vehicle for 30 minutes. Immunocytochemical analyses for NF-κB p65 were performed, as described previously.
24 Each average ratio of the number of cells with nuclear p65 staining to that of total cells per microscopic field was analyzed.
Statistical Analyses
All results were expressed as mean ± SD. The values were processed for statistical analyses (Mann-Whitney U test). Differences were considered statistically significant at P < 0.05.
Results
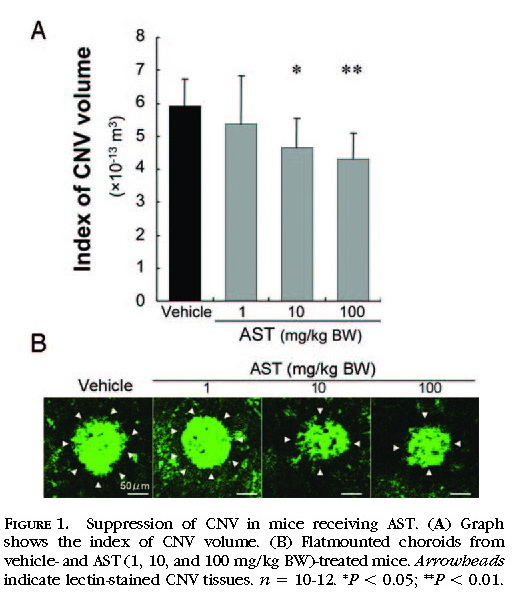
Suppression of CNV in Mice Receiving AST
The index of CNV volume was measured to evaluate the effects of AST treatment on the development of CNV. CNV was significantly suppressed by treatment with AST. AST-treated mice, at the dose of 10 or 100 mg/kg BW, showed a significant d
ecrease in the index of CNV volume (464,738 ± 87,719 μm
3 for 10 mg/kg BW and 431,321 ± 77,338 μm
3 for 100 mg/kg BW) compared with vehicle-treated mice (591,283 ± 82,688 μm
3;
Figs. 1A 1B ).
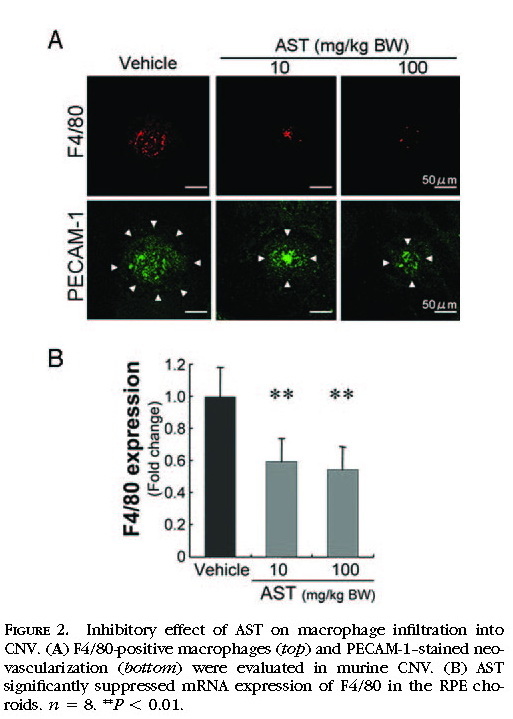
Suppression of Macrophage Infiltration by Treatment with AST
As the cellular mechanism in the pathogenesis of CNV, the infiltration of inflammatory cells, including macrophages, plays a critical role in the growth of CNV. We analyzed the infiltration of macrophages in murine CNV by immunohistochemistry
(Fig. 2A)and quantitative RT-PCR
(Fig. 2B)for the macrophage-specific antigen F4/80. Immunoreactivity for F4/80 was substantially lower in AST-treated mice at the dose of 10 or 100 mg/kg BW than in vehicle-treated mice
(Fig. 2A) . In the quantitative analyses using the real-time PCR, AST-treated mice at the dose of 10 or 100 mg/kg BW showed a significant decrease in the expression of F4/80 in the RPE–choroid complex compared with vehicle-treated animals (
P< 0.01;
Fig. 2B ).
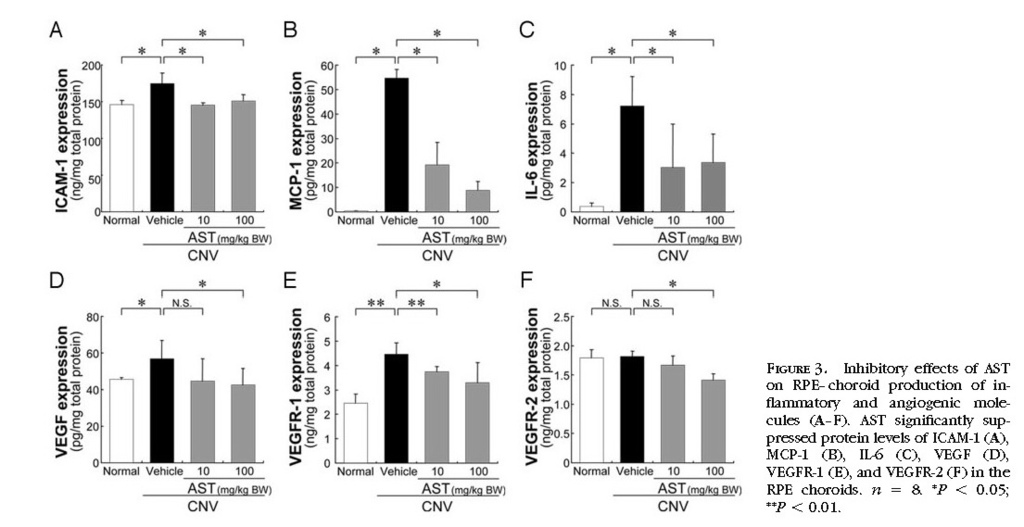
In Vivo Inhibition of Inflammatory and Angiogenic Molecules by the Treatment with AST
To determine whether AST treatment affects inflammatory and angiogenic molecules related to the pathogenesis of CNV, protein levels of ICAM-1, MCP-1, IL-6, VEGF, VEGFR-1, and VEGFR-2 in the RPE–choroid complex were analyzed by ELISA. RPE–choroid levels of ICAM-1, MCP-1, IL-6, VEGF and VEGFR-1 were significantly higher in mice with CNV than in age-matched healthy controls. AST treatment significantly suppressed protein levels of ICAM-1, MCP-1, IL-6, VEGF, VEGFR-1 and VEGFR-2
(Figs. 3A 3B 3C 3D 3E 3F) .
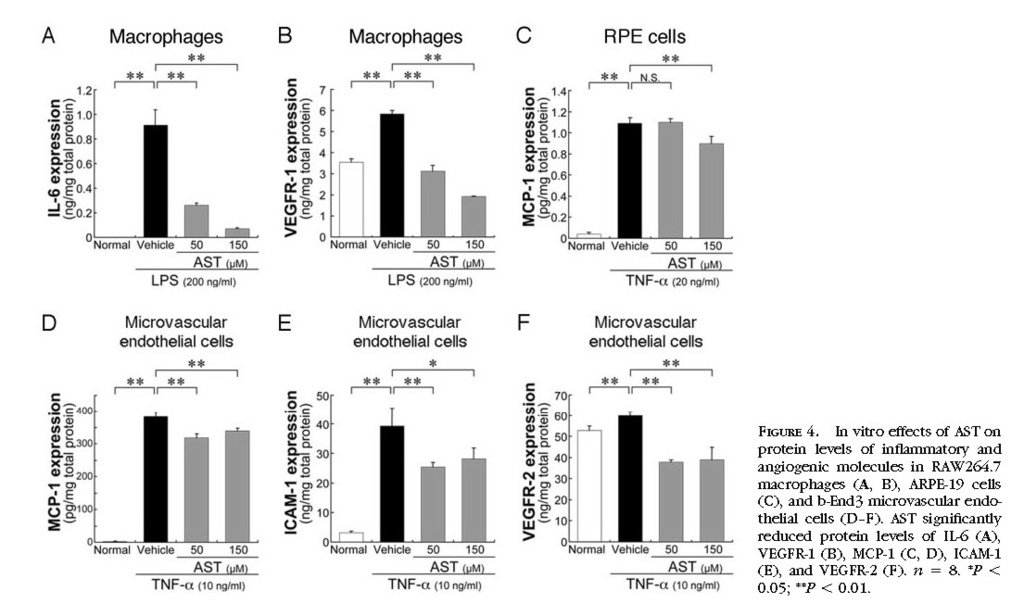
In Vitro Inhibition of Inflammatory and Angiogenic Molecules by Treatment with AST
To confirm in vivo effects of AST on choroidal inflammation and neovascularization, we further performed in vitro analyses. In RAW264.7 macrophages, AST treatment significantly (
P < 0.01) reduced the protein levels of LPS-induced IL-6
(Fig. 4A)and VEGFR-1
(Fig. 4B) . In ARPE-19 cells, AST application led to a significant (
P < 0.01) decrease in the protein levels of TNF-α–induced MCP-1
(Fig. 4C) . Similarly, in b-End3 cells, AST treatment significantly (
P < 0.01) reduced protein levels of MCP-1
(Fig. 4D) , ICAM-1
(Fig. 4E) , and VEGFR-2
(Fig. 4F) , all of which were induced by TNF-α. In contrast, VEGF production was not significantly changed by treatment with AST, either in RAW264.7 macrophages or in ARPE-19 cells (data not shown).
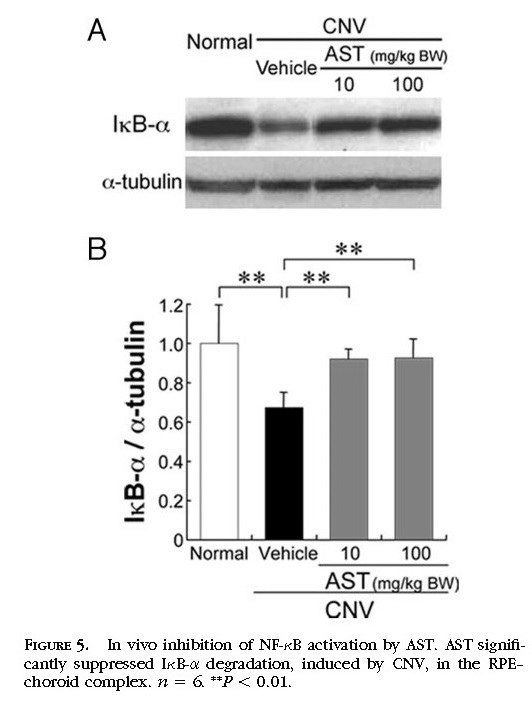
In Vivo Inhibition of NF-κB Activation by Treatment with AST
To define the signaling pathway involved in treatment with AST, we focused on NF-κB as an upstream transcriptional factor of inflammatory mediators and analyzed the protein level of IκB-α in vivo. In the murine RPE–choroid tissues, photocoagulation induced NF-κB activation, including IκB-α degradation
(Figs. 5A 5B) . Protein levels of IκB-α in the RPE–choroid complex were significantly (
P < 0.01) reduced 4 hours after photocoagulation compared with age-matched healthy controls. AST significantly (
P < 0.01) inhibited IκB-α degradation at 4 hours in the murine RPE–choroid complex
(Figs. 5A 5B) .
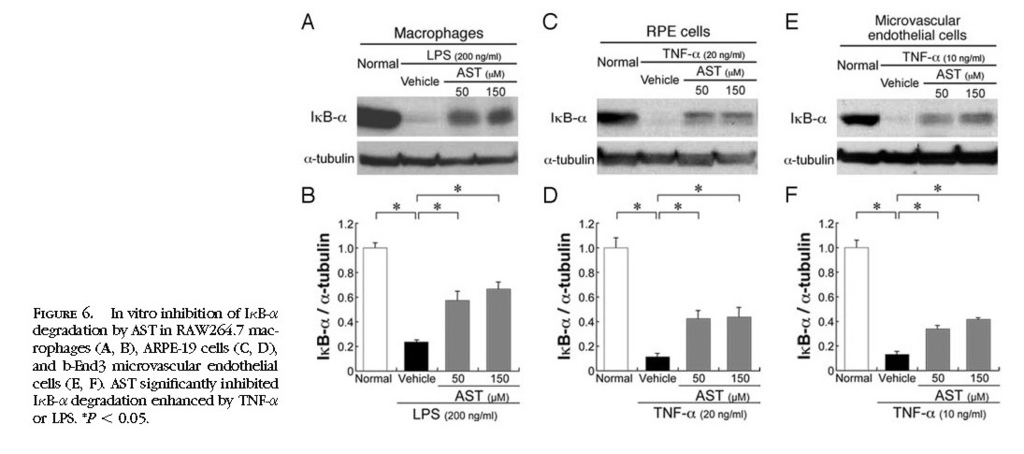
In Vitro Inhibition of NF-κB Activation by Treatment with AST
To further confirm in vivo effects of AST on NF-κB inhibition, we performed in vitro experiments using RAW264.7, ARPE-19, and b-End3 cells stimulated by LPS or TNF-α. AST significantly (
P < 0.05) inhibited IκB-α degradation enhanced by LPS in RAW264.7 cells
(Figs. 6A 6B)or by TNF-α in ARPE-19 cells
(Figs. 6C 6D)and b-End3 cells
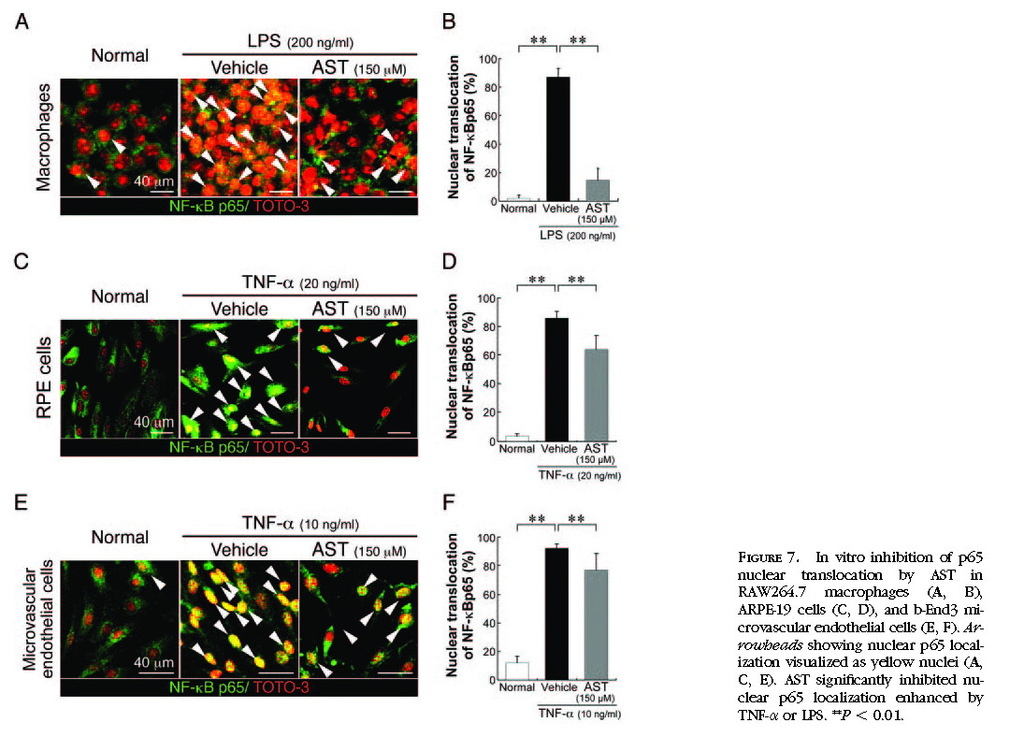
(Figs. 6E 6F) . Nuclear translocation of NF-κB p65, enhanced by TNF-α or LPS, was significantly (
P < 0.01) suppressed by the application of AST in RAW264.7 cells
(Figs. 7A 7B) , ARPE-19 cells
(Figs. 7C7D) , and b-End3 cells
(Figs. 7E 7F) .
Discussion
The present study reveals several important findings concerning the antipathogenic role of AST in the development of CNV. First, treatment with AST led to significant suppression of CNV
(Fig. 1) . Second, the cellular and molecular mechanisms in AST treatment included the inhibitory effects on macrophage infiltration into CNV
(Fig. 2)and inflammation-related molecules in the RPE–choroid complex
(Fig. 3)and in cultured macrophages, RPE cells, and microvascular endothelial cells
(Fig. 4) . Third, AST treatment resulted in the inhibition of NF-κB activation in vivo
(Fig. 5)and in vitro
(Figs. 6 7) .
Various observational
25 26 27 and interventional
28 29 studies showed the possibility of carotenoid consumption, including AST,
28 lutein,
25 26 29 and β-carotene,
27 for reducing the risk for AMD. Accordingly, the effect of carotenoid supplementation for preventing and treating AMD has recently been attracting attention. We have revealed that lutein is anti-inflammatory in preventing CNV in mice.
23 AST has higher antioxidant activity toward peroxyl radicals than lutein.
30 However, no data have been presented concerning the inhibitory effect of AST on CNV development. We have demonstrated for the first time that AST treatment led to the suppression of CNV
(Fig. 1) .
As cellular mechanisms for suppressing CNV by the treatment with AST, the present data showed that AST application led to significant suppression of macrophage infiltration
(Fig. 2) . Pharmacologic depletion of macrophages, which accumulated in murine CNV tissues,
31resulted in significant suppression of murine CNV.
7 9 One of roles of macrophages in the development of CNV is to promote neovascularization by secreting VEGF at the lesion where RPE and vascular endothelial cells produce MCP-1 for macrophage recruitment.
32Our present data are compatible with the recent data showing that AST application led to in vivo suppression of inflammatory cell infiltration in the rodent model of endotoxin-induced uveitis
19 20 and in the rabbit model of atherosclerosis.
33
As molecular mechanisms for suppressing CNV, the present data showed the AST-induced suppression of various inflammation-related molecules, including VEGF, VEGFR-1, VEGFR-2, IL-6, ICAM-1, and MCP-1, which were upregulated after the induction of CNV
(Fig. 3) . Previous reports concerning the molecular mechanisms underlying CNV generation showed VEGF as a promoting mediator.
5 31 34 Macrophages infiltrating into CNV are a rich source of VEGF. VEGFR-1 is expressed in inflammatory leukocytes, including macrophages.
35 The AST-induced decrease in RPE–choroid VEGF and VEGFR-1, seen in the present study
(Fig. 3) , is compatible with and is explained at least in part by the suppression of VEGF-secreting and VEGFR-1-bearing macrophage infiltration. Indeed, our in vitro experiments
(Fig. 4)showed no remarkable effects on VEGF production either in macrophages or in RPE cells after AST treatment (data not shown). Additionally, we performed in vitro experiments showing that IL-6 levels in macrophages, MCP-1 levels in RPE cells, and ICAM-1, MCP-1, and VEGFR-2 levels in vascular endothelial cells were significantly reduced by AST
(Fig. 4) . Recently, we have shown that CNV formation is mediated by IL-6 receptor signaling.
14Several in vivo experiments with genetically altered mice demonstrated a significant contribution of adhesion molecules and chemotactic factors, including ICAM-1
10 and MCP-1,
36 both of which are required for macrophage infiltration. VEGF-mediated endothelial cell mitogenic activity was shown to depend on VEGFR-2.
37 Collectively, the presently observed suppression of CNV by treatment with AST is likely attributable to the inhibition of multiple inflammatory steps, including MCP-1–induced migration and ICAM-1-dependent adhesion of macrophages, subsequent macrophage-derived VEGF and IL-6 secretion, and VEGFR-2 expression in endothelial cells.
Because NF-κB is suggested to induce the expression of inflammation-related molecules, we investigated the role of NF-κB in the development of CNV
(Figs. 5 6 7) . After IκB phosphorylation and degradation caused by various stimuli, NF-κB p65/p50, capable of entering the nucleus and binding the κB sequence, promotes the transcription of target genes including ICAM-1, MCP-1, and IL-6.
38 Recently, we have clarified the critical role of NF-κB in the development of CNV, showing the NF-κB inhibition with an inhibitor of p65 nuclear translocation (dehydroxymethylepoxyquinomicin) led to significant suppression of experimental CNV.
23 In the present study, AST inhibited the activation of NF-κB by suppressing IκB degradation
(Figs. 5 6)and subsequent p65 nuclear translocation
(Fig. 7)in the RPE–choroid in vivo
(Fig. 5)and in macrophages, RPE cells, and microvascular endothelial cells
(Figs. 6 7) . Collectively, our data suggest the importance of AST capable of inhibiting NF-κB activation.
Anti-VEGF therapy is applied for the treatment of AMD complicated by CNV.
39 Because the therapeutic intervention for blocking VEGF tends to be limited to the advanced stage, an alternative early treatment is thought to be required, targeting inflammation as an antecedent event leading to neovascularization. Epidemiologic risk factors for AMD include age, smoking, cardiovascular diseases such as atherosclerosis, and nutrient status.
25 40 41 42It is reasonable to intervene modifiable risk factors, such as nutrient status for the prevention of AMD. Accordingly, our present data may provide molecular evidence of the potential validity of AST supplementation as a therapeutic strategy to suppress CNV.
These authors contributed equally to the work presented here and should therefore be regarded as equivalent authors.
Supported by a Grant-in-Aid (no. 19592039) for Scientific Research from the Japanese Ministry of Education, Culture, Sports, Science and Technology (SI).
Submitted for publication November 5, 2007; revised December 7, 2007; accepted February 15, 2008.
Disclosure: K. Izumi-Nagai, None; N. Nagai, None; K. Ohgami, None; S. Satofuka, None; Y. Ozawa, None; K. Tsubota, None; S. Ohno, None; Y. Oike, None; S. Ishida, None
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Corresponding author: Susumu Ishida, Laboratory of Retinal Cell Biology, Department of Ophthalmology, Keio University School of Medicine, 35, Shinanomachi, Shinjuku-ku, Tokyo 160-8582, Japan;
ishidasu@sc.itc.keio.ac.jp.




 留言列表
留言列表
 線上藥物查詢
線上藥物查詢 