


David J. Greenblatt , MD
Educational Objectives
Upon completion of this educational activity, participants should be able to:
- Explain the mechanisms by which grapefruit juice (GFJ) may produce pharmacokinetic interactions with certain prescription drugs.
- Evaluate medications being taken by patients and discuss with their physicians which drugs, if any, may be of concern when taken with GFJ.
- Consult with patients' physicians in selecting drugs within a given class that may be safely taken with GFJ, with little or no risk of a drug interaction.
- Caution patients and other health care professionals regarding sources of information that may be incomplete or inaccurate.
Twenty years have passed since the first report of a pharmacokinetic drug interaction involving grapefruit juice (GFJ) and the prescription calcium channel antagonist agent, felodipine.1 Since this time, hundreds of papers have appeared in the medical and scientific literature reporting on the mechanisms and consequences of pharmacokinetic interactions between GFJ and a number of prescription medications.2-12 The topic has found its way into many secondary and tertiary sources, including Web sites, pharmacy compendia, and drug information guides. The lay media has also given prescription drug interactions with GFJ extensive coverage. The principal areas of focus will be the physiologic and biochemical mechanisms of such interactions, research methods to assess interactions with GFJ, approaches to evaluating the clinical importance of these events, and potential sources of bias and misinterpretation.
The Science of DDIs
A pharmacokinetic drugrug interaction (DDI) involves a situation in which one drug (the "perpetrator") changes the clearance and plasma concentration of another drug (the "substrate" or "victim").13-17 A pharmacokinetic DDI may or may not mean that the perpetrator changes the clinical effects of the victimhat depends on whether the change in plasma concentration of the substrate victim is sufficient to cause an alteration in the effects of the drug on the patient. Many DDIs that are statistically significant are also clinically unimportant. Possible reasons for this are (1) the magnitude of the interaction (ie, the change in plasma levels of the victim drug) is small enough that it cannot be detected clinically; or (2) the therapeutic index of the victim drug is large. DDIs are of most concern in clinical practice when either the magnitude of the interaction is large or the therapeutic index of the victim is narrow.
Not all DDIs are pharmacokinetic. Pharmacodynamic DDIs may occur when the perpetrator and victim act on the same pharmacologic target, but do not change each other's plasma levels or pharmacokinetics. An example would be the partial reversal of the sedative effects of alcohol by caffeine. DDIs involving GFJ are thought to be exclusively pharmacokinetic rather than pharmacodynamic.
Induction Versus Inhibition
DDIs typically (although not always) occur through the perpetrator's effect on the metabolic mechanisms mediating the clearance of the substrate victim (Table 1). Metabolic inhibition implies that the perpetrator impairs the biotransformation of the victim.18-22 The result is that plasma concentrations of the victim become higher, raising the possibility (although not the certainty) that therapeutic effects may be enhanced, or that toxicity may result. With metabolic induction, the perpetrator causes an increase in the clearance of the substrate victim, and plasma levels of the victim fall to lower levels.17,21,22 The concern in this case is that the therapeutic effect of the substrate victim will be diminished, or it may become ineffective altogether.

Inhibition and induction do not simply represent the same biologic process going in opposite directions. They are fundamentally different processes having different mechanisms. Inhibition represents a direct chemical effect of an inhibitor on a metabolic enzyme. The effect is rapid onset and is relatively rapidly reversed when the perpetrator drug is removed. Inhibition can be studied in vitro using liver cell homogenates, and a drug's inhibitory potency can be quantitated using measures such as an inhibition constant (Ki) or a 50% inhibitory concentration (IC50). In contrast, induction is a more complex process in which the perpetrator (the inducer), acting as a transcriptional activator via binding to cellular receptors, causes increased synthesis of metabolic protein.23-26 This transcriptional and synthetic cascade requires several days or more to be complete and is equally slow to be reversed when the inducing agent is removed. In vitro study of metabolic induction requires a system in which protein synthetic mechanisms are intactamely, cell cultures of intact hepatocytes. Unlike inhibition, no well-established quantitative metric exists to depict a drug's potency as an inducer.
Design of DDI Studies
Clinical studies of DDIs have become a standard component of the drug development process and, likewise, provide important information on the therapeutic benefits and risks of already marketed drugs that may be coadministered in clinical practice. Standards for the conduct of DDI studies have evolved over the years, and reasonable consensus now exists among the regulatory and academic communities as to how studies should be designed and executed.13-16,27-29
The principal outcome measure in DDI studies is the area under the plasma concentration-time curve (AUC) for the substrate victim drug. In singledose DDI studies, AUC represents the total area from time 0 to infinity. If the victim is given in multiple doses such that steady-state is attained, AUC is the segmental area over a dosing interval at steady-state. The typical study has a crossover design, in which AUC for the substrate victim is measured on 2 different occasions in the same subject: once in the control condition (AUC0), without coadministration of the candidate perpetrator; and on a second occasion (AUCI) during coadministration of the perpetrator. For each study participant, an AUC ratio is calculated as follows:
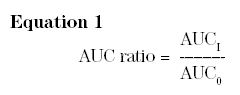
The individual AUC ratios are then aggregated across subjects. The appropriate statistical method for aggregation is a matter of some controversy. The most straightforward, transparent, and understandable approach is a calculation of the arithmetic mean and standard deviation of the individual ratios.30 Statistical significance of the interaction is then easily tested using student's t test, comparing the arithmetic mean ratio with 1.0. The FDA guidance on DDI studies requires a nonstraightforward approach, the basis for which lies in statistical theory. The FDA mandates analysis of DDI study data as if it were a bioequivalence studyhe geometric mean of individual AUC ratios is calculated, along with the 90% confidence interval (CI).31 The interaction is deemed statistically significant if 1.0 is incorporated within the boundaries of the 90% CI. Unfortunately, the geometric mean ratio always underestimates the true arithmetic mean, and the FDA aggregation method therefore underestimates the actual magnitude of the interaction.30 Nonetheless, the 2 statistical methods generally yield the same "overall conclusion" about the drug interactions, and either way, an AUC ratio equal to 1.0 indicates no interaction; >1.0 indicates inhibition; and <1.0 indicates induction.
Clinical Importance of a DDI: Statistical Versus Clinical Significance
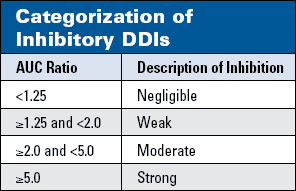
The bigger the AUC ratio, the more likely it is that a DDI involving metabolic inhibition will be clinically important. "Default" descriptors, generated through experience and general consensus, are consistent with this concept (Table 2). Still, interpretation of the outcome of a DDIhether the aggregate AUC ratio indicates statistical significanceust be guided by the following principle:
A pharmacokinetic DDI study does not, by itself, allow a conclusion as to whether a DDI is clinically important. This judgment requires supplemental data on the exposure-response relationship for the substrate victim drug.
As an example, assume a DDI study shows a mean AUC ratio of 1.5. Whether this is clinically important depends entirely on the exposureresponse properties of the victim. If the victim is warfarin, a ratio of 1.5 is very likely to be important, and the DDI would require either avoidance of the drug combination, or reduction in the daily dosage of warfarin. If the victim is penicillin, a ratio of 1.5 is unlikely to be important.
Application of these principles to studies of drug interactions with GFJ is discussed in sections that follow.
Biochemical Mechanisms of Interactions with GFJ
The Role of CYP3A
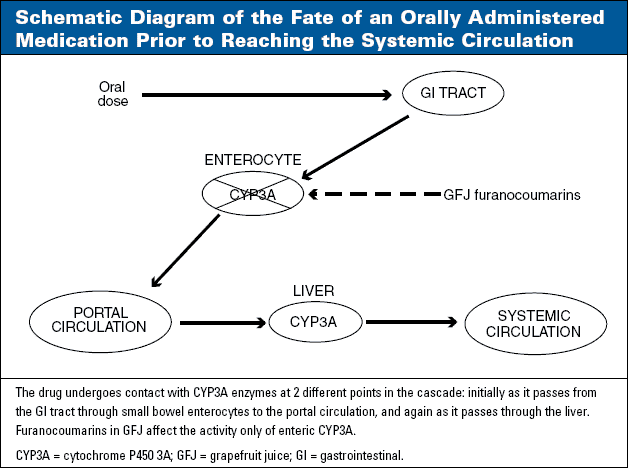
Among the human cytochrome P450s (CYPs), CYP3A (referring to CYP3A4 and CYP3A5) is of greatest quantitative importance in the human liver, and is responsible, either entirely or in part, for the metabolic clearance of at least half of the drugs currently used in clinical practice.32,33 CYP3A isoforms also are the only CYP enzymes present in clinically meaningful amounts in enterocyte cells found in the mucosa of the proximal small bowel, which is the site of absorption of most drugs. An orally administered drug that is a substrate for metabolism for CYP3A therefore is sequentially exposed to CYP3A initially during absorption through small bowel enterocytes and again during passage through the liver (Figure 1). Significant metabolism at either or both of these sites produces incomplete oral bioavailability due to presystemic extraction or first-pass metabolism.
How Does GFJ Cause Drug Interactions?
Drug interactions with GFJ are caused by inhibition of the activity of CYP3A located in enterocyte cells in the small bowel mucosa. Among the many natural substances present in GFJ, the furanocoumarins are established as the class of compounds responsible for CYP3A inhibition (Figure 2).34-42 Bergamottin is the most abundant of the furanocoumarins in GFJ; however, bergamottin is a weak inhibitor and probably contributes to drug interactions only to a small extent. The most important inhibitor appears to be 6',7'-dihydroxybergamottin (DHB), which is a potent inhibitor of CYP3A. Also of importance as potent CYP3A inhibitors are a series of dimers, termed spiroesters or paradisins, formed by linkage of DHB with itself, or DHB with bergamottin.

Customary exposure to GFJ (in the range of 8-12 oz per day of regular-strength GFJ) causes inhibition only of enteric CYP3Aepatic CYP3A is not affected (Figure 1). The mode of CYP3A inhibition is termed mechanism-based orirreversible.43-46 This implies that GFJ, as the perpetrator, does not need to be present in the gastrointestinal (GI) tract at the time the substrate victim is given to produce an interaction with the victim. Prior exposure to GFJ inactivates the enzyme, and recovery occurs at a rate consistent with the normal turnover and regeneration of CYP3A.47-52 This process has a half-life of about 24 hours.
Necessary Conditions for a Drug Interaction with GFJ
Based on the above, along with other evidence in the medical and scientific literature, the circumstances under which GFJ may produce a pharmacokinetic drug interaction are now established.
- The substrate drug must be biotransformed by CYP3A. GFJ does not produce drug interactions with substrates metabolized by CYP enzymes other than CYP3A. As examples, GFJ does not interact with warfarin53 (CYP2C9) or theophylline54 (CYP1A2).
- The substrate drug must be given orally. Except with unrealistically high doses,55 GFJ affects only those CYP3A enzymes present in GI tract mucosal cells. CYP3A in the liver is not affected. A number of studies demonstrate that clearance of intravenously administered substrates is not impaired by GFJ, even when GFJ reduces clearance and elevates AUC for the same substrates given orally.56-60
- The substrate must ordinarily undergo presystemic extraction by enteric CYP3A following oral dosage. Substrates that are metabolized by CYP3A, but do not undergo presystemic extraction (ie, have oral bioavailability close to 100%) are not affected by GFJ. Examples include alprazolam61 and quinidine.62 For substrates (such as midazolam) that are subject to presystemic extraction by a combination of hepatic and enteric CYP3A,63 GFJ will impair only the component attributable to enteric CYP3A.
An important conclusion is that all 3 of these conditions are necessary for a drug interaction with GFJ to be possible.
Drug Interactions with GFJ
Table 3 summarizes the current state of knowledge on the topic of prescription drug interactions with GFJ. Interaction categories have been generated based on 2 considerations. The first point of consideration is the magnitude of the interaction. With GFJ as the potential inhibitor (perpetrator), Equation 1 becomes:
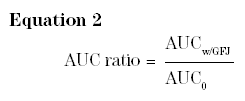
The larger the AUC ratio, the greater the likelihood of a clinically important interaction (Table 2). AUC ratios >5.0 generally indicate clinically important interactions, whereas AUC ratios <2.0 generally (but not always) suggest low or no risk of a clinically important interaction. The second consideration is the exposure-response relationship for the specific victim. In Table 3, the actual AUC ratios have been interpreted in this context.
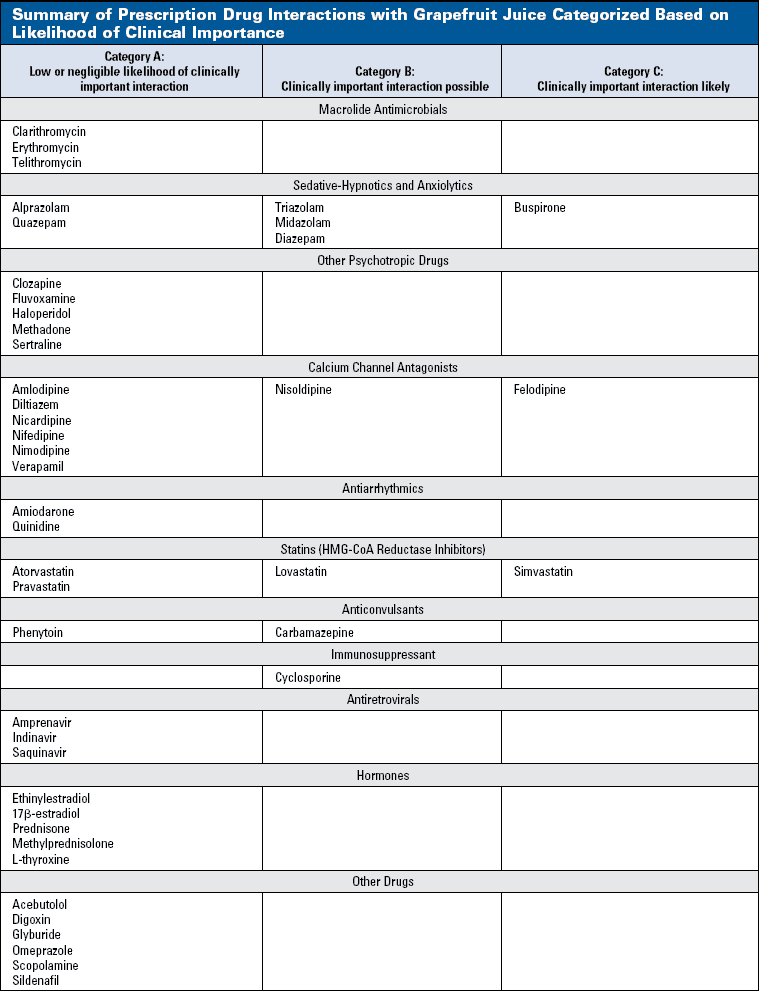
The 3 interaction categories are:
- Low or negligible likelihood of a clinically important interaction. Patients taking drugs in this category can drink GFJ with no reason to worry about a drug interaction.
- A drug interaction with GFJ is possible. GFJ does not need to be avoided in patients taking substrate drugs in Category B, but patients and physicians should be aware that the drug's clinical effects may be increased.
- Significant likelihood of a clinically important drug interaction with GFJ. For safety purposes, GFJ should be avoided, or another drug having similar therapeutic effects (ie, a drug in the same class) with a lower likelihood of an interaction should be substituted.
The drugs categorized and listed in Table 3 include agents that have been evaluated in drug interaction studies with GFJ and are commonly used in clinical practice. Sparsely used drugs (such as antimalarial and antiparasitic agents) are not included. Also not included are drugs withdrawn from clinical use (such as terfenadine and cisapride), or drugs not available in the United States (such as the beta-blockers talinolol and celiprolol). Finally, we have assumed typical and "realistic" consumption of GFJuch as 8 to 10 oz of regular-strength GFJ dailys opposed to larger quantities and/or double-strength GFJ as used in some research studies.
Not all original references have been cited. The reader is referred to a comprehensive review article,10 as well as a Web site providing extensive documentation on interactions of drugs with foods and nutrients (www.cop.ufl.edu/fdic/index.php).
Some Cases in Point
Several drugs for which the AUC ratio (Equation 2) falls below 2.0 warrant some additional discussion.
Cyclosporine
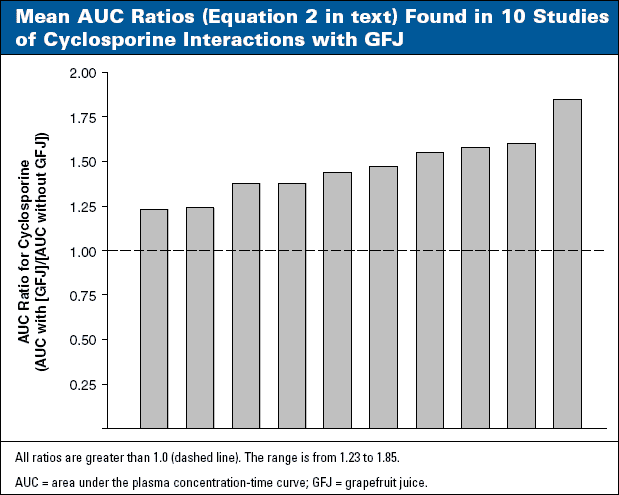
A number of studies of cyclosporine interactions with GFJ have been published. AUC ratios in these studies consistently fall below 2.0 and range from 1.23 to 1.85 (Figure 3). Nonetheless, cyclosporine has been placed in Category B (Table 3), because patients receiving cyclosporine generally constitute a "vulnerable" population and because of the narrow therapeutic range of cyclosporine and the potentially hazardous toxicity.
Carbamazepine
Only one study of a carbamazepine and GFJ has been published.64 This shows an AUC ratio of 1.4. Nonetheless, carbamazepine has been listed in Category B due to the narrow therapeutic index and the risk of toxicity.
Erythromycin
The macrolide antimicrobial erythromycin has the pharmacologic property of delaying cardiac repolarization and prolonging the electrocardiographic QT interval.65,66 Epidemiologic studies have demonstrated an increased risk of sudden death among patients taking erythromycin, compared with controls receiving amoxicillin.67 The risk associated with erythromycin appeared to be increased by concurrent use of strong CYP3A inhibitors, such as ketoconazole and itraconazole. A single study of GFJ and erythromycin in healthy volunteers showed an AUC ratio of 1.48, indicating "weak" inhibition (Table 2).68 We have listed erythromycin in Category A, because no evidence exists to indicate that an interaction of this magnitudef it applied to patients taking erythromycinould present any hazard of serious cardiac arrhythmias beyond that associated with erythromycin alone.
Data Sources to Be Wary of
The entries in Table 3 are based on pharmacokinetic data derived from controlled studies in humans. This, in principle, is the only source of biomedical "truth" with respect to the potential risks and benefits of GFJ use by patients concurrently taking prescription medications. Other types of data are available in the literature, however. These other data sources cannot be assumed to represent biomedical truth, although such assumptions may be invited or encouraged by the authors, and actually be accepted as truth in some secondary and tertiary sources and by the lay media.
Case Reports
Case reports describe anecdotal events occurring in individual patients. An example of a typical case report (hypothetical only) is as follows:
A patient with a lipid disorder has been taking a statin derivative for 3 months. The patient then starts drinking GFJ, 8 oz daily with breakfast. Two weeks after starting GFJ, the patient complains of muscle weakness and soreness. The authors of the report speculate that an interaction of GFJ with the statin caused an elevation of plasma concentrations to toxic levels, thereby explaining the patient's symptoms.
It is generally recognized by the scientific community that anecdotal case reports do not constitute evidence of cause-and-effect.69-71 Such reports may represent a "signal" of a phenomenon that deserves further investigation via controlled studies, but do not by themselves prove causality. Problems with the above hypothetical report include: (1) the author's description is the only representation of clinical events. No medical records or other documents are available, and there is no way to verify the validity of the report; (2) the muscle weakness and soreness are subjective descriptions by the patient, with no laboratory or other verification; (3) muscle symptoms may occur for reasons other than an adverse effect of a statin; (4) statins may cause muscle symptoms in patients not taking GFJ; (5) plasma levels of the statin before and after GFJ exposure were not available to verify that a drug interaction actually occurred.
The consequences and clinical risks (if any) of coadministration of statinsr other prescription drugsith GFJ must be deduced from controlled human studies (as in Table 3), as opposed to observational reports on individual patients.
Studies in Experimental Animals
The outcome of GFJrug interaction studies done in experimental animals are sometimes interpreted as predictive of the outcome of coadministration of GFJ with the same drug in humans. This extrapolation is not appropriateo known animal model exists that allows valid prediction of drug interactions in humans.
The typical experimental model involves rats or other rodent species. The predictive limitations of the rat model include the following72-75: (1) the dosage of GFJ (on a weight-normalized basis) generally is much higher than the typical human exposure; (2) CYP3A enzymes in rats are different from the CYP3A enzymes in humans; (3) the substrate victim drug may not be metabolized to the same extent by rat CYP3A compared with human CYP3A; (4) the relative distributions of enteric and hepatic CYP3A differ between rats and humans, as does the role of enteric CYP3A in presystemic extraction.
The conclusion is that no assumptions can be made regarding GFJrug interactions in humans on the basis of experimental animal studies.
In Vitro Models
In vitro models, usually based on microsomal preparations of human liver, now are widely used in the process of drug development and in academic settings to identify and anticipate DDIs that may occur in vivo. In the case of small molecular entities used as prescription drugs, the in vitro modelsespite significant limitationsave proved useful in identifying drug combinations that might interact in humans, as well as excluding combinations unlikely to interact.13,14,18,19,27-29,32,76-85 In the case of nutrients and natural products, however, the in vitro models have very limited predictive value. We have termed this the "in vitron vivo disconnect."12 Many fruit beveragesncluding cranberry and pomegranateave been identified as reversible CYP3A inhibitors in vitro,86,87 but only GFJ interacts importantly with CYP3A in humans. It appears that irreversible (mechanismbased) inhibition of CYP3A is a requisite for fruit beverages to cause clinically important drug interactions, and only GFJ furanocoumarins produce this type of inhibition. The conclusion is that no assumptions can be made regarding the clinical applicability of in vitro studies of CYP inhibition by fruit beverages.
Secondary and Tertiary Sources
All secondary and tertiary sourcesncluding this articlenvolve a "filtration" or interpretation of primary research data by the authors or compilers of the document. The interpretation will depend on the training, experience, and biases of the document author(s). Different secondary/tertiary sources may interpret the same primary data differently. Busy health care professionals may understandably not have the opportunity to evaluate primary research data themselves and may come to rely on secondary or tertiary sources for information on potential drug interactions with GFJ. Still, the filtration process should be kept in mind, and a return to the primary data may sometimes be needed to resolve conflicting or uncertain information.
Transporter-Based Drug Interactions
All drug interactions with GFJ discussed so far involve effects of GFJ on drug metabolism via CYP3A. Recently, a mechanistically different category of interaction has come to light, involving drug transport rather than drug metabolism. The organic acid transporter polypeptides (OATPs) are a class of uptake transporter proteins responsible for facilitating GI absorption of a limited number of drug entities.88-92 Components of GFJs well as components of orange juicenhibit the activity of OATP, such that absorption of affected drugs will be reduced by coadministration of GFJ or orange juice.93-95 Furanocoumarins are not the components responsible for the interaction.96
Affected drugs are few in number.93 The antihistamine fexofenadine is the only drug available in the United States for which GFJ substantially reduces absorption.97-99 The magnitude of the interaction depends on the volume of GFJ ingested, and the time between fexofenadine dosage and GFJ exposure. The interaction can be minimized by separating the ingestion times by 2 hours or more.
The scientific literature on DDIs via this newly recognized mechanism will need to be followed, and recommendations updated, as additional data become available.
Comment
Medical and scientific research on the topic of drug interactions with GFJ (and other nutrients) should have the ultimate objective of protecting and improving public health. If there is a realistic risk of an undesired outcome from combining GFJ with a specific prescription drug, health care professionals and patients should be informed of this so that appropriate modifications and precautions can be taken. If a realistic risk does not exist, however, warning to that effect does notadvance public health. Unnecessary warnings may create concern and anxiety, impair adherence to needed pharmacologic treatment plans, and deter consumption of a nutrient that may have health benefits or otherwise enhance quality of life. The scientific community should put biases aside and look for the proper balance of caution.
另外補充一些三總臨床藥學部提供的整理:
在發現葡萄柚汁對felodipine有交互作用後,有一連串的研究探討葡萄柚汁對其他CCB的影響。腸內CYP系統對於CCB成員的代謝程度有相當大的差異,因此絕對生體可用率也有差異性,葡萄柚汁對於口服生體可用率較低的CCB影響較大。研究發現,併用felodipine和葡萄柚汁會使血壓下降更多,有心跳增加以及增加血管擴張的現象,其他dihydropyridine CCBs,nisoldipine以及nicardipine也發現類似的交互作用。另外,nimodipine和nitrendipine跟葡萄柚汁併服會增加50%到100%的生體可用率。Amlodipine和nifedipine的生體可用率較不受影響,但是亦會增加約20%到30%。屬於nondihydropyridine的diltiazem和verapamil雖然經由CYP3A4,和葡萄柚汁併用不會影響它們的生體可用率。
最開始葡萄柚汁與felodipine有交互作用被報導後,很快就發現對於其他柑橘類,例如柳丁汁,不會有類似的作用,因為葡萄柚汁含有特別會引起此交互作用的物質。葡萄柚汁中主要flavonoid是naringin,使葡萄柚具有特殊芳香以及苦味,naringin是Cytochrome p-450 enzymes的受質,不存於其他柑橘類或是果汁中。Nonflavonoid中的6',7 dihydroxybergamottin以及bergamottin具有抑制CYP3A4的作用。然而,葡萄柚汁對CYP3A4的作用並不是單獨只由這些成分引起,臨床上葡萄柚汁和CYP3A4的作用是很多成分共同影響的結果。
研究亦發現,在大部分的情況下使用一般新鮮未經加工的葡萄柚汁一杯(250ml),就會產生與CCBs的交互作用,而且不是只有藥物和葡萄柚汁同時服用才會產生交互作用。例如,葡萄柚汁對felodipine的交互作用,在喝下葡萄柚汁二十四小時後才服用felodipine仍然有30%的影響。葡萄柚汁與藥物交互作用的時間長短跟腸道中產生新的酵素來恢復藥物原有的代謝所需要的時間相關,一般建議葡萄柚汁和CCBs至少要相隔二十四小時以上,因此建議服用CCBs的病人儘量避免喝葡萄柚汁才是比較安全的。
References
- Bailey DG, Spence JD, Edgar B, Bayliff CD, Arnold JMO. Ethanol enhances the hemodynamic effects of felodipine. Clin Invest Med. 1989;12(6):357-362.
- Bailey DG, Arnold JMO, Spence JD. Grapefruit juice and drugs. How significant is the interaction? Clin Pharmacokinet. 1994;26(2):91-98.
- Ameer B, Weintraub RA. Drug interactions with grapefruit juice. Clin Pharmacokinet. 1997;33(2):103-121.
- Bailey DG, Arnold JMO, Spence JD. Grapefruit juice-drug interactions. Br J Clin Pharmacol. 1998;46(2):101-110.
- Fuhr U. Drug interactions with grapefruit juice. Extent, probable mechanism and clinical relevance. Drug Saf. 1998;18(4):251-272.
- Kane GC, Lipsky JJ. Drug-grapefruit juice interactions. Mayo Clin Proc. 2000;75(9):933-942.
- Greenblatt DJ, Patki KC, von Moltke LL, Shader RI. Drug interactions with grapefruit juice: an update. J Clin Psychopharmacol. 2001;21(4):357-359.
- Dresser GK, Bailey DG. The effects of fruit juices on drug disposition: a new model for drug interactions. Eur J Clin Invest. 2003;33(suppl 2):10-16.
- Bailey DG, Dresser GK. Interactions between grapefruit juice and cardiovascular drugs. Am J Cardiovasc Drugs. 2004;4(5):281-297.
- Mertens-Talcott SU, Zadezensky I, De Castro WV, Derendorf H, Butterweck V. Grapefruit-drug interactions: can interactions with drugs be avoided? J Clin Pharmacol. 2006;46(12):1390-1416.
- Paine MF, Oberlies NH. Clinical relevance of the small intestine as an organ of drug elimination: drug-fruit juice interactions. Expert Opin Drug Metab Toxicol. 2007;3(1):67-80.
- Farkas D, Greenblatt DJ. Influence of fruit juices on drug disposition: discrepancies between in vitro and clinical studies. Expert Opin Drug Metab Toxicol. 2008;4(4):381-393.
- Greenblatt DJ, von Moltke LL. Clinical studies of drug-drug interactions: design and interpretation. In: Pang KS, Rodrigues AD, Peter RM, eds. Enzyme and Transporter-Based Drug-Drug Interactions: Progress and Future Challenges. New York, NY: Springer; 2010:625-649.
- Farkas D, Shader RI, von Moltke LL, Greenblatt DJ. Mechanisms and consequences of drug-drug interactions. In: Gad SC, ed. Preclinical Development Handbook: ADME and Biopharmaceutical Properties. Philadelphia, PA: Wiley-Interscience; 2008:879-917.
- Greenblatt DJ, von Moltke LL. Drug-drug interactions: clinical perspectives. In: Rodrigues AD, ed. Drug-Drug Interactions. 2nd ed. New York, NY: Informa Healthcare; 2008:643-664.
- Greenblatt DJ, von Moltke LL. Sedative-hypnotic and anxiolytic agents. In: Levy RH, Thummel KE, Trager WF, Hansten PD, Eichelbaum M, eds. Metabolic Drug Interactions. Philadelphia, PA: Lippincott, Williams and Wilkins; 2000:259-270.
- Lin JH. CYP induction-mediated drug interactions: in vitro assessment and clinical implications. Pharm Res. 2006; 23(6):1089-1116.
- Venkatakrishnan K, von Moltke LL, Greenblatt DJ. Effects of the antifungal agents on oxidative drug metabolism in humans: clinical relevance. Clin Pharmacokinet. 2000;38(2):111-180.
- Venkatakrishnan K, von Moltke LL, Greenblatt DJ. Human drug metabolism and the cytochromes P450: application and relevance of in vitro models. J Clin Pharmacol. 2001;41(11):1149-1179.
- Venkatakrishnan K, von Moltke LL, Obach RS, Greenblatt DJ. Drug metabolism and drug interactions: application and clinical value of in vitro models. Curr Drug Metab. 2003;4(5):423-459.
- Pelkonen O, Turpeinen M, Hakkola J, Honkakoski P, Hukkanen J, Raunio H. Inhibition and induction of human cytochrome P450 enzymes: current status. Arch Toxicol. 2008;82(10):667-715.
- Zhou SF, Xue CC, Yu XQ, Li C, Wang G. Clinically important drug interactions potentially involving mechanism-based inhibition of cytochrome P450 3A4 and the role of therapeutic drug monitoring. Ther Drug Monit. 2007;29(6):687-710.
- Wang H, LeCluyse EL. Role of orphan nuclear receptors in the regulation of drug-metabolizing enzymes. Clin Pharmacokinet. 2003;42(15):1331-1357.
- Willson TM, Kliewer SA. PXR, CAR and drug metabolism. Nat Rev Drug Discov. 2002;1(4):259-266.
- Urquhart BL, Tirona RG, Kim RB. Nuclear receptors and the regulation of drug-metabolizing enzymes and drug transporters: implications for inter-individual variability in response to drugs. J Clin Pharmacol. 2007;47(5):566-578.
- Goodwin B, Redinbo MR, Kliewer SA. Regulation of CYP3A gene transcription by the pregnane X receptor. Annu Rev Pharmacol Toxicol. 2002;42:1-23.
- Huang SM, Strong JM, Zhang L, et al. New era in drug interaction evaluation: US Food and Drug Administration update on CYP enzymes, transporters, and the guidance process. J Clin Pharmacol. 2008;48(6):662-670.
- Bjornsson TD, Callaghan JT, Einolf HJ, et al. The conduct of in vitro and in vivo drug-drug interaction studies: a PhRMA perspective. J Clin Pharmacol. 2003;43(5):443-469.
- Huang SM, Temple R, Throckmorton DC, Lesko LJ. Drug interaction studies: study design, data analysis, and implications for dosing and labeling. Clin Pharmacol Ther. 2007;81(2):298-304.
- Greenblatt DJ. Preparation of scientific reports on pharmacokinetic drug interaction studies. J Clin Psychopharmacol. 2008;28(4):369-373.
- Steinijans VW, Hartmann M, Huber R, Radtke HW. Lack of pharmacokinetic interaction as an equivalence problem. Int J Clin Pharmacol Ther Toxicol. 1991; 29(8):323-328.
- Greenblatt DJ, He P, von Moltke LL, Court MH. The CYP3 family. In: Ioannides C, ed. Cytochrome P450: Role in the Metabolism and Toxicology of Drugs and Other Xenobiotics. Cambridge, United Kingdom: Royal Society of Chemistry; 2008:354-383.
- Guengerich FP. Cytochrome P-450 3A4: regulation and role in drug metabolism. Annu Rev Pharmacol Toxicol. 1999;39:1-17.
- Kakar SM, Paine MF, Stewart PW, Watkins PB. Dihydroxybergamottin contributes to the grapefruit juice effect. Clin Pharmacol Ther. 2004;75(6):569-579.
- Paine MF, Widmer WW, Hart HL, et al. A furanocoumarin-free grapefruit juice establishes furanocoumarins as the mediators of the grapefruit juice-felodipine interaction. Am J Clin Nutr. 2006;83(5):1097-1105.
- Edwards DJ, Bellevue FH 3rd, Woster PM. Identification of 6',7'-dihydroxybergamottin, a cytochrome P450 inhibitor, in grapefruit juice. Drug Metab Dispos. 1996;24(12):1287-1290.
- Fukuda K, Ohta T, Oshima Y, Ohashi N, Yoshikawa M, Yamazoe Y. Specific CYP3A4 inhibitors in grapefruit juice: furocoumarin dimers as components of drug interaction. Pharmacogenetics. 1997;7(5):391-396.
- Schmiedlin-Ren P, Edwards DJ, Fitzsimmons ME, et al. Mechanisms of enhanced oral availability of CYP3A4 substrates by grapefruit constituents. Decreased enterocyte CYP3A4 concentration and mechanism-based inactivation by furanocoumarins. Drug Metab Dispos. 1997;25(11):1228-1233.
- Guo LQ, Fukuda K, Ohta T, Yamazoe Y. Role of furanocoumarin derivatives on grapefruit juice-mediated inhibition of human CYP3A activity. Drug Metab Dispos. 2000;28(7):766-771.
- Ohta T, Maruyama T, Nagahashi M, et al. Paradisin C: a new CYP3A4 inhibitor from grapefruit juice. Tetrahedron. 2002;58:6631-6635.
- Girennavar B, Poulose SM, Jayaprakasha GK, Bhat NG, Patil BS. Furocoumarins from grapefruit juice and their effect on human CYP 3A4 and CYP 1B1 isoenzymes. Bioorg Med Chem. 2006;14(8):2606-2612.
- Guo LQ, Yamazoe Y. Inhibition of cytochrome P450 by furanocoumarins in grapefruit juice and herbal medicines. Acta Pharmacol Sin. 2004;25(2):129-136.
- Zhou S, Chan E, Lim LY, et al. Therapeutic drugs that behave as mechanism-based inhibitors of cytochrome P450 3A4. Curr Drug Metab. 2004;5(5):415-442.
- Fontana E, Dansette PM, Poli SM. Cytochrome p450 enzymes mechanism based inhibitors: common sub-structures and reactivity. Curr Drug Metab. 2005;6(5):413-454.
- Zhou S, Yung Chan S, Cher Goh B, et al. Mechanism-based inhibition of cytochrome P450 3A4 by therapeutic drugs. Clin Pharmacokinet. 2005;44(3):279-304.
- Silverman R. Mechanism-based enzyme inactivators. Methods Enzymol. 1995;249:240-283.
- Greenblatt DJ, von Moltke LL, Harmatz JS, et al. Time course of recovery of cytochrome p450 3A function after single doses of grapefruit juice. Clin Pharmacol Ther. 2003;74(2):121-129.
- Culm-Merdek KE, von Moltke LL, Gan L, et al. Effect of extended exposure to grapefruit juice on cytochrome P450 3A activity in humans: comparison with ritonavir. Clin Pharmacol Ther. 2006;79(3):243-254.
- Takanaga H, Ohnishi A, Murakami H, et al. Relationship between time after intake of grapefruit juice and the effect on pharmacokinetics and pharmacodynamics of nisoldipine in healthy subjects. Clin Pharmacol Ther. 2000;67(3):201-214.
- Lundahl J, Regardh CG, Edgar B, Johnsson G. Relationship between time of intake of grapefruit juice and its effect on pharmacokinetics and pharmacodynamics of felodipine in healthy subjects. Eur J Clin Pharmacol. 1995;49(1-2):61-67.
- Lilja JJ, Kivist� KT, Neuvonen PJ. Duration of effect of grapefruit juice on the pharmacokinetics of the CYP3A4 substrate simvastatin. Clin Pharmacol Ther. 2000;68(4):384-390.
- Takanaga H, Ohnishi A, Matsuo H, et al. Pharmacokinetic analysis of felodipine-grapefruit juice interaction based on an irreversible enzyme inhibition model. Br J Clin Pharmacol. 2000;49(1):49-58.
- Sullivan DM, Ford MA, Boyden TW. Grapefruit juice and the response to warfarin. Am J Health Syst Pharm. 1998;55(15):1581-1583.
- Fuhr U, Maier A, Keller A, et al. Lacking effect of grapefruit juice on theophylline pharmacokinetics. Int J Clin Pharmacol Ther. 1995;33(6):311-314.
- Lilja JJ, Kivist� KT, Backman JT, Neuvonen PJ. Effect of grapefruit juice dose on grapefruit juice-triazolam interaction: repeated consumption prolongs triazolam half-life. Eur J Clin Pharmacol. 2000;56(5):411-415.
- Rashid TJ, Martin U, Clarke H, Waller DG, Renwick AG, George CF. Factors affecting the absolute bioavailability of nifedipine. Br J Clin Pharmacol. 1995;40(1):51-58.
- Uno T, Ohkubo T, Sugawara K, et al. Effects of grapefruit juice on the stereoselective disposition of nicardipine in humans: evidence for dominant presystemic elimination at the gut site. Eur J Clin Pharmacol. 2000;56(9-10):643-649.
- Ducharme MP, Warbasse LH, Edwards DJ. Disposition of intravenous and oral cyclosporine after administration with grapefruit juice. Clin Pharmacol Ther. 1995;57(5):485-491.
- Kupferschmidt HH, Ha HR, Ziegler WH, Meier PJ, Kr鄣enbl S. Interaction between grapefruit juice and midazolam in humans. Clin Pharmacol Ther. 1995;58(1):20-28.
- Lundahl J, Regardh CG, Edgar B, Johnsson G. Effects of grapefruit juice ingestionharmacokinetics and haemodynamics of intravenously and orally administered felodipine in healthy men. Eur J Clin Pharmacol. 1997;52(2):139-145.
- Yasui N, Kondo T, Furukori H, et al. Effects of repeated ingestion of grapefruit juice on the single and multiple oral-dose pharmacokinetics and pharmacodynamics of alprazolam. Psychopharmacology. 2000;150(2):185-190.
- Min DI, Ku YM, Geraets DR, Lee H. Effect of grapefruit juice on the pharmacokinetics and pharmacodynamics of quinidine in healthy volunteers. J Clin Pharmacol. 1996;36(5):469-476.
- Tsunoda SM, Velez RL, von Moltke LL, Greenblatt DJ. Differentiation of intestinal and hepatic cytochrome P450 3A activity with use of midazolam as an in vivo probe: effect of ketoconazole. Clin Pharmacol Ther. 1999;66(5):461-471.
- Ketter TA, Post RM, Worthington K. Principles of clinically important drug interactions with carbamazepine. Part II. J Clin Psychopharmacol. 1991;11(5):198-203, 306-313.
- Nattel S, Ranger S, Talajic M, Lemery R, Roy D. Erythromycin-induced long QT syndrome: concordance with quinidine and underlying cellular electrophysiologic mechanism. Am J Med. 1990;89(2):235-238.
- Oberg KC, Bauman JL. QT interval prolongation and torsades de pointes due to erythromycin lactobionate. Pharmacotherapy. 1995;15(6):687-692.
- Ray WA, Murray KT, Meredith S, Narasimhulu SS, Hall K, Stein CM. Oral erythromycin and the risk of sudden death from cardiac causes. N Engl J Med. 2004;351(11):1089-1096.
- Kanazawa S, Ohkubo T, Sugawara K. The effects of grapefruit juice on the pharmacokinetics of erythromycin. Eur J Clin Pharmacol. 2001;56(11):799-803.
- Goldman SA. Limitations and strengths of spontaneous reports data. Clin Ther. 1998;20(suppl C):C40-C44.
- Greenblatt DJ, von Moltke LL. Interaction of warfarin with drugs, natural substances, and foods. J Clin Pharmacol. 2005;45(2):127-132.
- Venning GR. Validity of anecdotal reports of suspected adverse drug reactions: the problem of false alarms. Br Med J (Clin Res Ed). 1982;284(6311):249-252.
- Perloff MD, von Moltke LL, Greenblatt DJ. Ritonavir and dexamethasone induce expression of CYP3A and P-glycoprotein in rats. Xenobiotica. 2004;34(2):133-150.
- Warrington JS, von Moltke LL, Shader RI, Greenblatt DJ. In vitro biotransformation of sildenafil (Viagra) in the male rat: the role of CYP2C11. Drug Metab Dispos. 2002;30(6):655-657.
- Warrington JS, Greenblatt D, von Moltke LL. Role of CYP3A enzymes in the biotransformation of triazolam in the rat liver. Xenobiotica. 2004;34(5):463-471.
- Kotegawa T, Laurijssens BE, von Moltke LL, et al. In vitro, pharmacokinetic, and pharmacodynamic interactions of ketoconazole and midazolam in the rat. J Pharmacol Exp Ther. 2002;302(3):1228-1237.
- Volak LP, Greenblatt DJ, von Moltke LL. In vitro approaches to anticipating clinical drug interactions. In: Li AP, ed. Drug-Drug Interactions in Pharmaceutical Development. Hoboken, NJ: John Wiley & Sons; 2008:75-93.
- von Moltke LL, Greenblatt DJ, Schmider J, et al. In vitro approaches to predicting drug interactions in vivo. Biochem Pharmacol. 1998;55(2):113-122.
- Youdim KA, Zayed A, Dickins M, et al. Application of CYP3A4 in vitro data to predict clinical drug-drug interactions; predictions of compounds as objects of interaction. Br J Clin Pharmacol. 2008;65(5):680-692.
- Galetin A, Ito K, Hallifax D, Houston JB. CYP3A4 substrate selection and substitution in the prediction of potential drug-drug interactions. J Pharmacol Exp Ther. 2005;314(1):180-190.
- Zhang ZY, Wong YN. Enzyme kinetics for clinically relevant CYP inhibition. Curr Drug Metab. 2005;6(3):241-257.
- Ito K, Brown HS, Houston JB. Database analyses for the prediction of in vivo drug-drug interactions from in vitro data. Br J Clin Pharmacol. 2004;57(4):473-486.
- Ohno Y, Hisaka A, Suzuki H. General framework for the quantitative prediction of CYP3A4-mediated oral drug interactions based on the AUC increase by coadministration of standard drugs. Clin Pharmacokinet. 2007;46(8):681-696.
- Brown HS, Galetin A, Hallifax D, Houston JB. Prediction of in vivo drug-drug interactions from in vitro data: factors affecting prototypic drug-drug interactions involving CYP2C9, CYP2D6 and CYP3A4. Clin Pharmacokinet. 2006;45(10):1035-1050.
- Bachmann KA. Inhibition constants, inhibitor concentrations and the prediction of inhibitory drug-drug interactions: pitfalls, progress and promise. Curr Drug Metab. 2006;7(1):1-14.
- Obach RS, Walsky RL, Venkatakrishnan K, et al. The utility of in vitro cytochrome P450 inhibition data in the prediction of drug-drug interactions. J Pharmacol Exp Ther. 2006;316(1):336-348.
- Ngo N, Yan Z, Graf TN, et al. Identification of a cranberry juice product that inhibits enteric CYP3A-mediated first-pass metabolism in humans. Drug Metab Dispos. 2009;37(3):514-522.
- Farkas D, Oleson LE, Zhao Y, et al. Pomegranate juice does not impair clearance of oral or intravenous midazolam, a probe for cytochrome P450-3A activity: comparison with grapefruit juice. J Clin Pharmacol. 2007;47(3):286-294.
- Hagenbuch B, Gui C. Xenobiotic transporters of the human organic anion transporting polypeptides (OATP) family. Xenobiotica. 2008;38(7-8):778-801.
- Poirier A, Funk C, Lav� T, Noe J. New strategies to address drug-drug interactions involving OATPs. Curr Opin Drug Discov Devel. 2007;10(1):74-83.
- DuBuske LM. The role of P-glycoprotein and organic anion-transporting polypeptides in drug interactions. Drug Saf. 2005;28(9):789-801.
- Kim RB. Organic anion-transporting polypeptide (OATP) transporter family and drug disposition. Eur J Clin Invest. 2003; 33(suppl 2):1-5.
- Mikkaichi T, Suzuki T, Tanemoto M, Ito S, Abe T. The organic anion transporter (OATP) family. Drug Metab Pharmacokinet. 2004;19(3):171-179.
- Greenblatt DJ. Analysis of drug interactions involving fruit beverages and organic anion-transporting polypeptides. J Clin Pharmacol. 2009;49(12):1403-1407.
- Kirby BJ, Unadkat JD. Grapefruit juice, a glass full of drug interactions? Clin Pharmacol Ther. 2007;81(5):631-633.
- Glaeser H, Bailey DG, Dresser GK, et al. Intestinal drug transporter expression and the impact of grapefruit juice in humans. Clin Pharmacol Ther. 2007;81(3):362-370.
- Bailey DG, Dresser GK, Leake BF, Kim RB. Naringin is a major and selective clinical inhibitor of organic anion-transporting polypeptide 1A2 (OATP1A2) in grapefruit juice. Clin Pharmacol Ther. 2007;81(4):495-502.
- Dresser GK, Bailey DG, Leake BF, et al. Fruit juices inhibit organic anion transporting polypeptide-mediated drug uptake to decrease the oral availability of fexofenadine. Clin Pharmacol Ther. 2002;71(1):11-20.
- Dresser GK, Kim RB, Bailey DG. Effect of grapefruit juice volume on the reduction of fexofenadine bioavailability: possible role of organic anion transporting polypeptides. Clin Pharmacol Ther. 2005;77(3):170-177.
- Banfield C, Gupta S, Marino M, Lim J, Affrime M. Grapefruit juice reduces the oral bioavailability of fexofenadine but not desloratadine. Clin Pharmacokinet. 2002;41(4):311-318.
David J. Greenblatt, MD, is Professor and Chairman of the Department of Pharmacology and Experimental Therapeutics; Professor of Psychiatry, Medicine, and Anesthesia; Director of the Clinical Pharmacology Program, Tufts University School of Medicine and Tufts Medical Center; Associate Program Director, Tufts Medical Center Clinical/Translational Research Center, Boston, Massachusetts. Dr. Greenblatt discloses that he is a consultant to the Florida Department of Citrus, Lakeland, Florida. The editorial staff of Pharmacy Times and the staff of the Office of Continuing Professional Education have no relevant affiliations or financial relationships to disclose.


 留言列表
留言列表
 線上藥物查詢
線上藥物查詢 