

Author Information
1 Preventive Cardiology, Sterling Rock Falls Clinic, Sterling, Illinois, USA
2 University of Illinois College of Medicine, Department of Family and Community Medicine, Peoria, Illinois, USA
3 Southern Illinois University School of Medicine, Department of Family and Community Medicine, Springfield, Illinois, USA
Correspondence: Dr Peter P. Toth, Director of Preventive Cardiology, 101 East Miller Rd, Sterling Rock Falls Clinic, Sterling, IL 61081, USA.
1. Introduction
Cholesterol and lipids such as phospholipids and triglycerides play critical roles within the histological organization and intermediary metabolism of eukaryotes. Cholesterol is a precursor to steroid hormones and bile salts. Triglycerides are a storage form of energy, while their constituent fatty acids serve as oxidative substrates for a variety of tissues, such as skeletal muscle and myocardium. Cell membranes and the surface coat of lipoproteins are made up of phospholipids. Lipids are also involved in the regulation of cell membrane fluidity, the modulation of enzyme activity and intracellular signalling systems. Sterols and fats are produced endogenously and can also be absorbed from a wide array of dietary sources. The regulation of lipid metabolism is understandably complex. Abnormalities in lipid metabolism give rise to a very large number of disease states; among the most important of these is atherosclerosis.
Atherosclerosis is widely prevalent throughout the world. Dyslipidaemia, hypertension, insulin resistance and diabetes mellitus, age, cigarette smoking, obesity and physical inactivity are among the most important risk factors for developing atherosclerosis and its clinical sequelae, including myocardial infarction (MI), ischaemic stroke, peripheral arterial disease and lower extremity amputation, and death. The identification and treatment of dyslipidaemia is a high clinical priority in cardiovascular medicine in both the primary and secondary prevention settings.[1,2] An atherogenic milieu promotes endothelial dysfunction and increased systemic inflammatory tone, as well as a pro-oxidative and prothrombotic state. These abnormalities promote the transmigration, subendothelial trapping and oxidative modification of atherogenic lipoproteins.[3,4] Oxidized lipoproteins and a variety of cytokines activate lipid scavenging by macrophages resident within the subendothelial space, promoting the formation of foam cells, fatty streaks and atheromatous plaques.[5,6]
This article focuses on lipoprotein metabolism, the role of dyslipidaemia in risk for atherosclerosis and cardiovascular disease, and current approaches to managing specific derangements in a fasting lipid profile (obtained 10–12 hours after the last meal). A variety of guidelines are available to help define specific target levels for each component of the lipid profile depending upon a given patient's risk for cardiovascular events.[7-9] The lipid management guidelines for Europe and the US are currently being revised and were not yet available at the time this article went to print. Consequently, the focus is on therapeutic options rather than the attainment of specific target levels for particular lipoproteins.
2. Lipoprotein Metabolism
2.1 Chylomicrons, Low-Density Lipoproteins and Very-Low-Density Lipoproteins
Cholesterol, monoglycerides, diglycerides, free fatty acids and phospholipids available from both dietary and biliary sources are absorbed from micelles in the intestinal lumen by translocator proteins located within the brush border of jejunal enterocytes. Inside the jejunocyte cholesterol and lipid are packaged together with phospholipids and apolipoprotein (apo) B48 to form chylomicrons. Chylomicrons are the largest lipoproteins and are released into the perimesenteric lymphatic system. Chylomicrons access the central circulation through the thoracic duct. Lipoprotein lipase (LPL) hydrolyses triglycerides in chylomicrons, a process that yields chylomicron remnant particles (figure 1). Chylomicron remnants are cleared from the circulation by the low-density lipoprotein (LDL) receptor-like protein. Thus, chylomicrons facilitate delivery of dietary lipid and cholesterol to the liver.
Fig. 1
The liver secretes very-low-density lipoprotein (VLDL), a lipoprotein comprised of triglycerides, cholesterol esters and apo B100. LPL hydrolyses the triglycerides in VLDL. As the amount of triglyceride in these particles decreases, the VLDL is progressively converted into intermediate-density lipoprotein (IDL) and LDL. All of these lipoproteins are atherogenic. The free fatty acids released by lipolysis can be either used as substrates by skeletal muscle or myocardium, or can be taken up by adipose tissue and then reassimilated into triglyceride for storage. LDL particles are concentrated with cholesterol and cholesterol esters and depleted of triglycerides. LDL is not directly secreted from hepatocytes; rather, it is produced by VLDL catabolism. During chylomicron and VLDL lipolysis, apolipoproteins in their phospholipid surface coats (apo AI, apo AII, apo CII and others) are released and can be used to form high-density lipoprotein (HDL) in serum. HDL particles can also be secreted de novo from hepatocytes and jejunal enterocytes.
Because triglycerides and cholesterol esters are hydrophobic, serum VLDL remnants and LDL particles function as delivery vehicles of cholesterol and oxidizeable substrate to peripheral tissues, including blood vessel walls. Prospective cohort investigations from around the world consistently demonstrate a continuous and graded relationship between serum levels of LDL cholesterol (LDL-C) and risk for atherosclerotic disease.[11-14] LDL and VLDL remnants not taken up by peripheral tissues can be cleared from the circulation by hepatic lipoprotein receptors. Therapies that upregulate the expression of hepatic LDL receptors decrease the risk for atherosclerosis because they reduce serum levels of atherogenic lipoproteins. When it comes to LDL-C, a general rule of thumb is ‘lower is better’. The goal for LDL-C is risk stratified, such that the higher the risk, the lower the goal. Risk can be estimated with the Framingham risk model or other means by which to estimate 10-year risk for coronary heart disease (CHD) events.[15] According to the National Cholesterol Education Program (NCEP), high-risk patients (e.g. established CHD, diabetes, peripheral arterial disease, symptomatic carotid artery disease, abdominal aortic aneurysm or a 10-year Framingham risk score >20%) should have an LDL-C target of <100 mg/dL (2.6 mmol/L). Among patients with CHD, it is a therapeutic option to reduce LDL-C to <70 mg/dL (1.8 mmol/L).[16,17]
2.2 High-Density Lipoproteins
Unlike other lipoproteins which are atherogenic, the HDLs are unique in that they appear to be vasculoprotective and antiatherogenic. HDL cholesterol (HDL-C) constitutes 20–30% of total serum cholesterol. HDL particles appear to oppose atherogenesis by inhibiting endothelial cell adhesion molecule and selectin expression, stimulating endothelial nitric oxide and prostacyclin production (both vasodilators and antagonists of platelet activation), inhibiting endothelial cell apoptosis, decreasing platelet aggregability and inhibiting LDL oxidation, among other mechanisms.[18] HDL promotes reverse cholesterol transport (RCT), a series of enzymatic reactions that stimulate the mobilization of intracellular cholesterol from macrophage foam cells and transport the cholesterol back to the liver for elimination as bile salts or biliary cholesterol[19] (figure 1). RCT has been validated experimentally in both animal and human models.[20-22] The protein cargo of HDL (i.e. its ‘proteosome’) consists of up to 75 different proteins and enzymes. The proteosome of HDL particles can vary in response to metabolic conditions and a large number of genetic polymorphisms, and these differences influence its functionality. The HDL proteosome includes lipid-modifying enzymes (e.g. lecithin cholesteryl acyltransferase, cholesteryl ester transfer protein), oxidation-reduction enzymes (paraoxonase, platelet-activating factor acetyl hydrolase, glutathione peroxidase), acute phase reactants (serum amyloid A, fibrinogen), apolipoproteins and immunity factors (complement proteins), among many others.[23,24]
With some exceptions (i.e. polymorphisms such as apo A-IMilano and apo A-IParis, among others), in prospective epidemiological and case-control studies conducted throughout the world, high HDL-C levels have been found to be protective against the development of CHD, ischaemic stroke and premature mortality.[13,25-27] A low level of HDL-C (i.e. <40 mg/dL [1.0 mmol/L] in men and <50 mg/dL [1.3 mmol/L] in women) is an independent risk factor for the development of CHD and for cardiovascular morbidity and mortality.[18] In general, patients with familial hypoalphalipoproteinaemia (low HDL) have an increased risk for premature CHD, while patients with familial hyperalphalipoproteinaemia are relatively resistant to atherosclerotic disease. An HDL >60 mg/dL (1.55 mmol/L) is a ‘negative’ risk factor and is assigned a ‘−1’ in Framingham risk scoring. It is advisable to raise HDL-C in patients with low baseline levels of this lipoprotein. The NCEP has not promulgated treatment targets for HDL because a target is difficult to discern from currently available studies. The American Diabetes Association supports target levels of >40 mg/dL for diabetic men and >50 mg/dL for diabetic women. A European Consensus Panel supports a goal of >40 mg/dL in high-risk patients and those with metabolic syndrome (‘insulin resistance syndrome’).[28]
2.3 Triglycerides
Recent meta-analyses suggest that hypertriglyceridaemia is an independent risk factor for cardiovascular disease.[29,30] The Framingham study showed that, as serum levels of triglycerides increase, there is a continuous increase in risk for CHD events in both men and women.[31] Hypertriglyceridaemia is a defining feature of insulin resistance and the metabolic syndrome. Hypertriglyceridaemia is also encountered in patients with familial combined hyperlipidaemia, chylomicronaemia, dysbetalipoproteinaemia, diabetes, as well as LPL-deficiency and hepatic lipase-deficiency states. Among patients with CHD and a history of an acute syndrome who are receiving HMG-CoA reductase inhibitor (statin) therapy, serum triglyceride levels >150 mg/dL (1.7 mmol/L) are associated with increased morbidity and mortality compared with patients with triglyceride levels <150 mg/dL.[32] Hypertriglyceridaemia can arise from excess ingestion of dietary fat, impaired capacity for triglyceride catabolism and disposal, or increased hepatic biosynthesis of triglyceride (e.g. high carbohydrate diet).
Hypertriglyceridaemia is frequently observed in patients with insulin resistance and hepatic steatosis. In patients with insulin resistance, the capacity of insulin to inhibit hormone-sensitive lipase in visceral adipocytes is decreased. Hormone-sensitive lipase hydrolyses triglycerides to free fatty acids. As the rate of free fatty acid release increases, the portal circulation and hepatic parenchyma are exposed to excess fatty acid. Once in the hepatocyte, fatty acids can be reassimilated into triglyceride and packaged into VLDL (thereby potentiating hypertriglyceridaemia), can be oxidized as fuel in the mitochondrial matrix (β-oxidation) or shunted toward gluconeogenesis (potentially exacerbating hyperglycaemia). If these systems are inadequate to the task of disposing of excess fatty acid, then the patient is vulnerable to the development of steatosis. When triglycerides are severely elevated (>500 mg/dL [5.65 mmol/L]), patients are at risk for developing pancreatitis. Among patients with triglycerides >500 mg/dL, reducing triglycerides is the first priority of therapy. In patients with serum triglycerides >200 mg/dL (2.26 mmol/L), the secondary priority of therapy after LDL-C reduction is the reduction of non-HDL-C. Non-HDL-C is defined as total cholesterol minus HDL-C. Like LDL-C, non-HDL-C targets are risk stratified and are computed by adding 30 to the LDL-C target. Non-HDL-C is a surrogate measure of apo B100 and total atherogenic lipoprotein burden in serum since it includes all atherogenic lipoproteins (VLDL, IDL, LDL, lipoprotein(a), as well as remnants). There is speculation that, at least in the next iteration of NCEP guidelines, non-HDL-C may become the primary target of therapy.
Some of the toxicity attributable to elevated triglycerides is that they impair lipoprotein metabolism. As triglycerides increase in serum, there is increased transfer of triglyceride mass from VLDL into LDL and HDL particles by the enzyme cholesteryl ester transfer protein (figure 1). Triglyceride enrichment renders LDL and HDL particles better targets for lipolysis by hepatic lipase. Hepatic lipase activity results in the production of increased numbers of small dense LDL particles, and stimulates the catabolism and progressive reduction in serum levels of HDL, resulting in an atherogenic lipid profile. Triglyceride reduction is an important component of any therapeutic effort to raise HDL-C.
3. Drug Therapy for Dyslipidaemia
3.1 HMG-CoA Reductase Inhibitors (Statins)
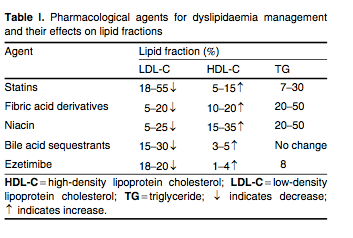
The statins are reversible, competitive inhibitors of HMG-CoA reductase, which is the rate-limiting step for cholesterol biosynthesis. Statins are first-line therapy for the treatment of elevated LDL-C. By reducing cholesterol biosynthesis and decreasing the size of the intracellular pool of available cholesterol, the statins stimulate the upregulation of the cell surface LDL receptors which promote the clearance of atherogenic apo B100-containing lipoproteins (VLDL, VLDL remnants, IDL and LDL). The statins also decrease hepatic VLDL secretion and lower serum levels of triglycerides, and increase apo A-I expression and HDL secretion. The statins stimulate hepatic HDL production by functioning as weak peroxisomal proliferator-activated receptor (PPAR)-α agonists.[33] Average changes in serum lipid levels for the various lipid-lowering drug classes are summarized in table I.
A large number of statin outcome trials have established that lowering LDL-C results in significant reductions in multiple ‘hard’ cardiovascular endpoints, including MI, stroke and death in primary[34,35] and secondary cohorts,[36,37] as well as in patients with diabetes,[38] hypertension[39] or heightened systemic inflammation,[40] as measured by serum high-sensitivity C-reactive protein levels exceeding 2.0 mg/L. As shown in JUPITER (see table II for a list of trial acronyms), statins benefit men and women, Blacks and Hispanics, and the elderly.[41] Statin therapy decreases the frequency and severity of angina pectoris and claudication,[36] decreases the frequency of coronary and peripheral revascularization,[40] and stabilizes or even reverses atherosclerotic plaque progression.[42,43] In the secondary prevention setting, lower LDL-C levels correlate with larger reductions in cardiovascular endpoints[44,45] and greater likelihood of coronary atherosclerotic plaque regression.[42,43] In addition, patients given higher doses of statins experience fewer acute cardiovascular events and require less frequent revascularization than patients given lower doses of these drugs.[46-48]

The Cholesterol Treatment Trialists Collaboration evaluated 14 prospective clinical trials using statin therapy and showed that for every 39 mg/dL (1 mmol/L) decrease in serum LDL-C with statin therapy over a mean treatment period of 5 years, the following reductions in outcomes were observed: 24% in MI or coronary death; 12% in all-cause mortality; 19% in coronary mortality; 24% in need for revascularization; and 17% in fatal/nonfatal stroke.[49] Similar results for these endpoints were observed among patients with diabetes.[50] For patients admitted to hospital with an acute coronary syndrome, it is standard of care to initiate statin therapy, irrespective of baseline lipid profile. It is recommended that in these patients the statin be initiated concomitant with aspirin, β-blockade and nitroglycerin. For patients with CHD or a CHD risk equivalent whose baseline LDL-C is below 100 mg/dL (2.6 mmol/L), LDL-C should be reduced at least 30% with statin therapy.[16] The specific statin and dose are chosen based on the patient's level of risk (low, moderate, moderately high, high or very high) and the percentage of LDL lowering needed.[51] The LDL-C reducing capacity of the statins can be summarized as follows: rosuvastatin 45–63% (5–40 mg/day); atorvastatin 26–60% (10–80 mg/day); simvastatin 26–47% (10–80 mg/day); lovastatin 21–42% (10–80 mg/day); fluvastatin 22–36% (10–20 mg/day); and pravastatin 22–34% (10–80 mg/day). Each doubling of the statin dose yields an additional 6% reduction, on average, in serum LDL-C (the so-called ‘rule of 6s’).
Despite a risk for myopathy and hepatotoxicity that is generally <1%, statins are safe and have a very large benefit to risk ratio.[51-53] Unfortunately, approximately 50% of patients discontinue their statin after only 1 year.[54] It is important for patients to understand why they are receiving statin therapy, and the importance of remaining compliant should be emphasized at every office follow-up visit. A statin should not be discontinued unless serum transaminase levels exceed three times the upper limit of normal. Elevations in serum transaminase levels are frequently mild and transient, and do not necessarily represent liver toxicity. Such patients may also have underlying hepatic steatosis. The most frequent reason cited for statin discontinuation is myalgia. Myalgia is observed in a community setting in 15–20% of patients treated with a statin.[52] When myalgia develops, it is important to rule out drug interactions, electrolyte disturbances, thyroid dysfunction and underlying fibromyalgia. In patients who develop myalgia, it is important to perform a pain assessment and localize whether the pain is occurring in joints or muscle. Occasionally the patient's perception of myalgia is really a manifestation of osteoarthritis-related pain. If the patient has myalgia, a lower dose of the same statin can be tried or switching to a different statin may be beneficial. There is no real documented difference in the incidence of myalgia with hydrophobic versus hydrophilic statins. Statin-induced myopathy/rhabdomyolysis has a large number of potential aetiologies.[52] Rhabdomyolysis is a medical emergency and represents diffuse skeletal muscle breakdown accompanied by large elevations in serum creatine kinase levels and myoglobinuria placing the patient at risk for acute renal failure.
The statins differ in their pharmacokinetic profiles due to differences in cytochrome P450 (CYP)-dependent metabolism and disposal. Lovastatin, pravastatin, fluvastatin and simvastatin have relatively short half-lives (1–4 hours) and should be taken in the evening in order to intercept the peak activity of HMG-CoA reductase, which occurs around midnight. Atorvastatin and rosuvastatin have long half-lives and can be taken at any time during the day or night. The coadministration of CYP3A4 inhibitors (macrolide antibiotics [erythromycin, clarithromycin], nefazodone, azole type antifungals [ketoconazole, itraconazole], HIV protease inhibitors, >1 quart [0.95 L] of grapefruit juice daily and ciclosporin) with lovastatin, simvastatin and atorvastatin is contraindicated as these statins are dependent on CYP3A4 metabolism, increasing risk for toxicity. The dose of simvastatin should not exceed 20 mg in patients receiving amiodarone or verapamil.
Statins do not completely eliminate the risk of CHD events as they often do not normalize all components of the lipid profile, especially in patients with metabolic syndrome, diabetes, severely depressed HDL-C or a variety of hypertriglyceridaemic states (familial combined hyperlipidaemia, hyperchylomicronaemia, severe LPL deficiency).[55] Patients with severely elevated LDL-C (familial hypercholesterolaemia, familial defective apo B100 and deficiency of 7-α-hydroxylase) may also be unable to normalize their lipid profile with statin monotherapy. Many of these patients will require the use of multiple lipid-lowering agents in order to achieve their risk-stratified NCEP target goals. Patients who cannot achieve their LDL-C target with therapeutic lifestyle change and statin therapy should be given adjuvant therapy with drugs such as nicotinic acid, ezetimibe or a bile acid sequestrant (BAS). Some patients with marked elevations in LDL-C who are high risk and require significant LDL-C reduction often need a statin, BAS and ezetimibe coupled with lifestyle modification in order to achieve their LDL-C target. In cases of severely elevated LDL-C (as encountered in homozygous familial hypercholesterolaemia where LDL-C can exceed 500 mg/dL [12.9 mmol/L]), aggressive combination drug therapy often has to be combined with LDL apheresis in specialized centres.
3.2 Bile Acid Sequestrants
The BAS (also known as bile-acid-binding resins) are anion exchange resins that bind bile acids in the gastrointestinal tract, thereby preventing their reabsorption across the terminal ileum and lowering their concentration in the enterohepatic circulation. The BAS reduce serum LDL-C by two mechanisms: (i) with reduced availability of bile acids, there is increased catabolism of cholesterol secondary to the upregulation of 7-α-hydroxylase, the rate-limiting enzyme for the conversion of cholesterol into bile acids; and (ii) with increased demand for intrahepatic cholesterol, hepatocytes increase their surface expression of LDL receptors, which promotes the clearance of apo B100-containing lipoproteins from plasma.
The capacity of BAS to beneficially impact cardiovascular outcomes has been demonstrated in the Lipid Research Clinics-Coronary Primary Prevention trial[56] and the Familial Atherosclerosis Treatment Study.[57] The BAS decrease serum LDL-C by 15–30% and increase HDL-C by 3–5% in a dose-dependent fashion. Unless a patient is statin intolerant, these drugs should be used in combination with a statin because BAS therapy stimulates HMG-CoA reductase activity in the liver, thereby increasing hepatic biosynthesis of cholesterol and offsetting the effects of the BAS over time. Therapy with a statin and colesevelam lowers LDL-C levels by up to 48%.[58-60] The BAS can be combined with ezetimibe in patients who are statin intolerant (e.g. myalgia, transaminases elevations in toxic range). BAS therapy is contraindicated in patients with baseline triglycerides >400 mg/dL (4.5 mmol/L) since they can exacerbate hypertriglyceridaemia. Colesevelam decreases haemoglobin A1c in patients with type 2 diabetes by approximately 0.4–0.6%.[61-63] The exact mechanism(s) of action by which BAS decrease glucose levels is not yet definitively known. The BAS impact activity of the nuclear transcription factor, farnesoid X receptor-α. This results in alterations in luminal bile acid composition, increases in the incretins (cholecystokinin and glucagon-like peptide-1) and reduces gluconeogenesis,[64] possibly mediated by improved insulin sensitivity.[65]
There are three BAS preparations available: colestyramine (cholestyramine; 4–24 g in two to three divided doses daily), colestipol (5–30 g in two to three divided doses daily) and colesevelam (1250 mg two to three times daily). Constipation, bloating and flatulence are relatively frequent adverse effects, although colesevelam has the lowest incidence of these complaints of the three BAS. The BAS bind all negatively charged molecules and drugs in the gastrointestinal lumen. Importantly, they can reduce the absorption of digitalis, thiazides, warfarin, phenobarbital, β-adrenoceptor antagonists (β-blockers), thyroxine, statins, fibric acid derivatives (fibrates) and ezetimibe. These medications should be taken 1 hour before or 4 hours after dosing BAS. The BAS reduce the absorption of fat-soluble vitamins. The BAS have no known direct systemic toxicity as they are not absorbed.
3.3 Ezetimibe
Ezetimibe inhibits cholesterol absorption at the intestinal brush border by binding to the Niemann-Pick C1-like 1 protein, a sterol transporter that takes up cholesterol and phytosterols (plant sterols) from the intestinal lumen into the jejunal enterocyte.[66,67] Ezetimibe decreases serum LDL-C levels by approximately 20%; in combination with statins, it has an additive LDL-C-lowering effect.[68-70] Ezetimibe also decreases triglycerides by up to 8% and raises HDL-C by 1–4%. Ezetimibe does not reduce the absorption of bile acids, steroid hormones (estrogens, progesterone) or fat-soluble vitamins such as vitamins A, D, E, or α- and β-carotenes. Ezetimibe can be used as primary therapy for LDL-C reduction in statin-intolerant patients and can be combined with BAS, fenofibrate, niacin and fish oils.
Ezetimibe is primarily used as an adjuvant therapy to statins for reducing elevated LDL-C. Ezetimibe is also available in fixed-dose combinations with simvastatin (Vytorin® 10 mg/10 mg, 10 mg/20 mg, 10 mg/40 mg, 10 mg/80 mg). Ezetimibe can be safely used in combination with other statins. Ezetimibe/simvastatin dosed at 10 mg/20 mg, 10 mg/40 mg or 10 mg/80 mg reduces LDL-C by 51%, 57% and 59%, respectively.[71] The LDL-C reduction with ezetimibe equates to approximately three statin titration steps (‘rule of 6s’). The addition of ezetimibe to a statin regimen substantially reduces the likelihood of having to titrate the statin. Statin-ezetimibe combination therapy is an important option for patients who do not tolerate moderate to high doses of statins or who refuse to comply with such doses. There is no clinical trial evidence that ezetimibe used independent of a statin beneficially impacts the risk for cardiovascular events. The ENHANCE study drew considerable controversy.[67] Among patients with familial hypercholesterolaemia, there was no statistically significant difference in rate of progression in carotid intima media thickness (CIMT) between groups treated with either simvastatin or the combination of simvastatin and ezetimibe. Although a lack of efficacy by ezetimibe was imputed by some,[72] it is also possible that the baseline CIMT between treatment groups was too thin to evaluate significant change over time.[73,74] In the Simvastatin and Ezetimibe Aortic Stenosis study, the combination of simvastatin and ezetimibe did not reduce progression of aortic valve stenosis, mortality or the need for aortic valve replacement in patients with aortic stenosis;[75] however, it did significantly reduce the need for coronary artery bypass grafting by 32% relative to placebo. The capacity of ezetimibe/simvastatin to reduce risk for cardiovascular events in patients with CAD is being studied in the IMPROVE-IT trial.[76]
3.4 Fibric Acid Derivatives
The fibrates are PPAR-α agonists and are useful for treating patients with hypertriglyceridaemia and mixed dyslipidaemia. These agents reduce serum triglycerides by 25–50% and raise HDL-C by 10–20%. The fibrates stimulate mitochondrial fatty acid uptake and β-oxidation, reduce triglyceride biosynthesis by inhibiting diacylglycerol acyltransferase-2 and decrease hepatic VLDL secretion.[30] Fibrates activate LPL by reducing levels of apo CIII (an inhibitor of LPL) and increasing levels of apo CII (an activator of LPL). The fibrates thus promote triglyceride hydrolysis in chylomicrons and VLDL. Fibrates increase HDL-C by both stimulating hepatic expression of apo A-I and reducing the loading of HDL with triglyceride, rendering it less vulnerable to lipolysis and catabolism by hepatic lipase. Fibrate therapy may be associated with an increase in serum LDL-C because of increased LPL-dependent conversion of VLDL to LDL. This can be dampened by simultaneously treating with an LDL-C-lowering therapy (i.e. statins, BAS or ezetimibe).
Gemfibrozil therapy is associated with reductions in cardiovascular events in both the primary and secondary prevention settings. In the HHS, 4081 men 40–55 years of age with a non-HDL-C level >200 mg/dL (5.2 mmol/L) were randomized to treatment with either gemfibrozil therapy (600 mg orally twice daily) or placebo.[77] The group treated with gemfibrozil experienced a 34% reduction in first-time CHD-related events. Among subjects with triglycerides >200 mg/dL (2.26 mmol/L) and HDL <42 mg/dL (1.08 mmol/L), the relative risk for CHD events decreased nearly 72% (statistically nonsignificant). In VA-HIT, men with CHD and low HDL-C (mean 31 mg/dL [0.8 mmol/L]) were randomized to receive either gemfibrozil (600 mg orally twice daily) or placebo over 5 years.[78] The group treated with gemfibrozil experienced a 22% reduction in the composite endpoint of all-cause mortality and nonfatal MI. Gemfibrozil treatment decreased the risk of stroke and transient ischaemic attacks by 31% and 59%, respectively, and decreased the need for carotid endarterectomy by 65%. Diabetic patients in VA-HIT experienced even greater benefit from gemfibrozil therapy, with reductions of 32% in the combined endpoint, 40% in stroke and 41% in CHD death.[79] VA-HIT was the first study to show a reduction in cardiovascular events with a lipid-lowering medication independent of changes in serum LDL-C.
The BIP trial was a secondary prevention trial comparing therapy with bezafibrate (400 mg/day) with placebo.[80] Patients were followed for an average of 6.2 years and the primary endpoints in the BIP trial were fatal or nonfatal MI and sudden death. Bezafibrate therapy reduced the risk for the primary composite endpoint by only 7.3%, which was not significant. However, among patients with a baseline serum triglyceride >200 mg/dL (2.26 mmol/L) and HDL <35 mg/dL (0.9 mmol/L), bezafibrate therapy decreased the composite endpoint significantly by 41%. Fibrates appear to benefit patients with high triglycerides and low HDL-C the most, although no study has yet been done to prospectively test this hypothesis. Bezafibrate is not available in the US. In the FIELD trial, fenofibrate therapy was compared with placebo in patients with type 2 diabetes. Although the study did not reach statistical significance in reducing the primary endpoint, fenofibrate was shown to decrease the risk of nonfatal MI (24%) and revascularization (21%), and to reduce the progression of microvascular disease, with a 38% reduction in lower extremity amputation, 31% reduction in the need for photocoagulation therapy for proliferative retinopathy and a 14% reduction in albuminuria.[81] The fibrates have not yet been shown to reduce mortality as an independent endpoint.
Like the statins, fibrates are associated with a low incidence of myopathy and mild elevations in serum transaminases. Fibrate therapy can increase risk for cholelithiasis, elevate serum homocysteine levels and raise prothrombin times by displacing warfarin from albumin binding sites. The three most commonly used fibrates are gemfibrozil (600 mg twice daily), fenofibrate (there are a variety formulations, both micronized and nonmicronized) and fenofibric acid (45 and 135 mg daily). Fenofibric acid is the only fibrate approved by the US FDA for use in combination with statins. Gemfibrozil significantly reduces the glucuronidation of statins and decreases their elimination.[82,83] The combination of gemfibrozil and statins is associated with increased risk for myopathy and hepatotoxicity. When used in combination with gemfibrozil, it is recommended that the doses for rosuvastatin and simvastatin not exceed 10 mg/day. When considering combination therapy with a statin and a fibrate for patients with mixed forms of dyslipidaemia, fenofibrate and fenofibric acid are safer choices since neither one of these fibrates inhibits statin glucuronidation. The ACCORD-LLA was designed to test the hypothesis of whether the addition of fenofibrate to simvastatin provides greater risk reduction compared with simvastatin therapy alone in patients with type 2 diabetes. Among patients with LDL-C 100 mg/dL (2.6 mmol/L), triglycerides 160 mg/dL (1.8 mmol/L) and HDL-C 39 mg/dL (1 mmol/L) on statin therapy, the addition of fenofibrate therapy did not reduce the risk for cardiovascular events or mortality.[84] However, in the group of patients with triglycerides >204 mg/dL (2.3 mmol/L) and HDL <34 mg/dL (0.88 mmol/L), there was a trend for a 31% reduction in the primary composite endpoint of cardiovascular events (p = 0.057). These results are consistent with those of other fibrate trials, such as HHS, BIP and FIELD. Of interest are the findings that there was no increase in risk for myopathy with combination therapy and that fenofibrate not only did not increase risk for adverse renal events, it also reduced both micro- and macroalbuminuria.
3.5 Niacin
Niacin, or nicotinic acid, is a form of vitamin B3. Niacin is a broad-spectrum lipid-modifying drug, which reduces LDL-C, triglycerides and lipoprotein a [Lp(a)], and increases HDL-C in a dose-dependent manner. Evidence of the efficacy of niacin in reducing cardiovascular events was first demonstrated in the Coronary Drug Project. Niacin therapy at 3.0 g/day was associated with a 26% reduction in risk of nonfatal MI and a 24% reduction in stroke compared with placebo in subjects with established CHD.[85] Mortality was reduced by 11%, which was not significant.
The mechanisms by which niacin exerts its effects are complex and as yet incompletely characterized. In patients with metabolic syndrome, niacin stimulates hepatic production of apo AI and HDL.[86] Niacin inhibits HDL particle uptake and catabolism by hepatocytes without reducing rates of RCT.[87] Both of these effects increase circulating levels of HDL. Niacin decreases hepatic VLDL and triglyceride secretion by the following mechanisms:
1. It decreases the flux of fatty acids from adipose tissue to the liver by inhibiting hormone-sensitive lipase activity. This effect is mediated by the receptor HM74 on the surface of visceral adipocytes and reduces the availability of substrate for hepatic triglyceride biosynthesis.[88]
2. It inhibits triglyceride formation within hepatocytes by inhibiting diacylglycerol acyltransferase, an enzyme that esterifies fatty acid to glycerol.
3. It reduces serum VLDL cholesterol and LDL-C concentrations by increasing the catabolism of apo B. By reducing serum triglyceride levels, niacin therapy increases LDL particle size and decreases LDL particle number.
Consequently, niacin beneficially impacts all components of the lipid profile.
Niacin can be added to a statin to treat combined hyperlipidaemia, especially if the HDL is low or Lp(a) is high. It must be emphasized that, as of yet, there are no prospective clinical trials demonstrating that Lp(a) reduction with niacin reduces the risk for cardiovascular events. Many lipidologists try to do this as something that is presumptively beneficial. In HATS, high-dose niacin (2–4 g) combined with simvastatin reduced cardiovascular morbidity and mortality by up to 90% compared with placebo. In addition, this form of combination therapy was associated with atheromatous plaque stabilization as assessed by quantitative coronary angiography over a 3-year follow-up period.[89] When added to statin therapy in patients with established CHD, extended-release (ER) niacin is associated with stabilization of CIMT; this is in contrast to those patients receiving statin monotherapy who experienced significant CIMT progression despite having a mean baseline LDL-C of 90 mg/dL (2.3 mmol/L) on statin monotherapy.[90] In an open-label extension of this trial with 57 patients, CIMT regression was highly correlated with the magnitude of HDL-C elevation on combination statin-niacin therapy.[91] The ARBITER 6-HALTS trial demonstrated significant regression in CIMT (mean baseline 0.90 mm) when patients with mean LDL-C 83 mg/dL (2.15 mmol/L) and HDL-C 43 mg/dL (1.1 mmol/L) on statin therapy are treated with 2.0 g of adjuvant ER niacin therapy over 14 months of follow-up.[92]
Niacin should be started at a low dose and slowly titrated upward (every 4–8 weeks) based on the results of follow-up lipid panels. Serum HDL levels can increase for up to 9 months at the 1.0 g/day dosage of niacin.[90] ER niacin induces the following changes in serum lipid levels across the dosing range of 500–2000 mg/day: LDL-C, 3–16% reduction; triglycerides, 5–32% reduction; and HDL-C, 10–24% elevation. The American Heart Association (AHA) recommends that ER niacin be the preferred choice of niacin for therapeutic use because of its purity, tolerability and low incidence of hepatotoxicity.[93] Niacin usage is limited by cutaneous flushing, a bothersome adverse effect. Flushing is the leading cause for discontinuation of therapy, estimated at 25–40% or more.[94,95] Flushing is mediated by prostaglandin D2 (PGD2), a potent vasodilator. PGD2 binds to DP1 receptors in the skin. ER niacin is associated with a lower frequency, intensity and duration of flushing than immediate-release niacin.[96-98] Limiting saturated fat intake for 2–3 hours before taking niacin can help reduce flushing because fat is a source of arachidonic acid, the substrate for cyclo-oxygenase. Aspirin is an irreversible inhibitor of cyclo-oxygenase, with higher doses of aspirin demonstrating greater efficacy than lower doses (325 mg compared with 80 mg or 160 mg).[99] It is also helpful to grind the aspirin and suspend it in clear juice, a manoeuvre that provides a much more robust, sudden absorption of aspirin across the gastric mucosa, and likely induces a more substantial and immediate level of inhibition of cyclo-oxygenase. On an anecdotal level, many providers advise the ingestion of applesauce with niacin, as this reduces flushing, possibly because the pectin slows rates of niacin absorption. Tredaptive® is a combination of niacin and laropiprant. Laropiprant is an antagonist of the DP1 receptor, inhibits cutaneous flushing and significantly improves the tolerability of niacin by over 50%.[100,101] This product is approved for use in the EU, but is not yet available in the US.
ER niacin is available as two combination formulations with lovastatin (Advicor®; 500 mg/20 mg, 1000 mg/20 mg and 2000 mg/40 mg) and simvastatin (Simcor®; 500 mg/20 mg, 750 mg/20 mg and 1000 mg/20 mg), with the two drugs inducing additive changes in serum lipids. The AIM-HIGH study is currently comparing the effects of ER niacin plus simvastatin with simvastatin monotherapy on the risk for cardiovascular events in patients with established CAD and atherogenic dyslipidaemia.[102]
3.6 Fish Oils
The cardiovascular benefits of omega-3 fatty acids (i.e. ‘fish oils’; eicosapentaenoic acid [EPA] and docosahexaenoic acid [DHA]) are well documented.[103-105] The AHA guidelines for secondary prevention recommend that patients consume two oily fish meals weekly or 1 g/day of EPA and DHA in capsule form, with up to 4 g/day in patients with hypertriglyceridaemia.[104] The omega-3 fatty acids reduce serum VLDL and triglycerides by reducing lipogenesis, increasing mitochondrial β-oxidation, increasing the catabolism of apo B and activating LPL.[30]
In the GISSI Prevenzione trial, use of omega-3 fatty acids (850 mg highly purified EPA + DHA per day) provided significant reductions in the risk for reinfarction and sudden death among patients who sustained an acute coronary syndrome prior to randomization.[106] JELIS randomized nearly 19 000 Japanese men and women with hypercholesterolaemia to statin therapy with or without 1800 mg/day of EPA.[107] The addition of EPA resulted in a 19% incremental reduction in major coronary events at 4.6 years of follow-up compared with statin monotherapy. A purified formulation (heavy metal contaminants, cholesterol, and saturated and oxidized fatty acids are removed) of EPA and DHA is available (Lovaza®) and is indicated at 4.0 g/day to treat serum triglyceride levels >500 mg/dL (5.65 mmol/L). At this dosage, Lovaza® can reduce serum VLDL and triglycerides by up to 42% and 45%, respectively.[108]
Patients with severe hypertriglyceridaemia can manifest with serum triglyceride levels in the thousands. These patients usually require complex combinations of lipid-lowering medication with fibrates, fish oils, statins, niacin and a low-fat diet. Patients with severe hypertriglyceridaemia can also benefit from treatment with the pancreatic lipase inhibitor orlistat, which significantly reduces the gastrointestinal absorption of fat. Among patients with hypertriglyceridaemia who also have diabetes, addition of the thiazolidenedione pioglitazone can reduce serum triglycerides significantly by relieving insulin resistance and promoting increased LPL activity.[109]
4. Conclusions
Dyslipidaemia is highly prevalent and commonly encountered throughout the world. The management of dyslipidaemia is a cornerstone in the prevention of primary and secondary cardiovascular events. The use of lipid-lowering therapies to reduce the risk for cardiovascular morbidity and mortality has been intensively studied for the last 3 decades, although questions remain. There is strong evidence to support the use of statins, fibrates, niacin, BAS and fish oils. Each of these drugs exerts its effects through distinct but often complementary mechanisms that can be used to rationally treat and beneficially impact abnormalities in lipid metabolism. The choice of drug(s) depends upon the specific disturbances in the lipid profile, and the patient's genetic and metabolic background. Mixed forms of dyslipidaemia frequently require the use of complex combinations of medications that can include three or more drugs from different classes. The combination of statins with niacin and fish oils has been studied prospectively in the HATS and JELIS trials, respectively. These combinations are effective and provide incremental benefit beyond statin monotherapy. The ACCORD-LLA trial supports the use of statin-fenofibrate therapy in diabetic patients with hypertriglyceridaemia and low HDL-C. Two drug combinations are also being evaluated prospectively in such trials as IMPROVE-IT (statin-ezetimibe) and AIM-HIGH (statin-niacin). Although no large prospective clinical trial outcome data are available to support the use of three or more drug combinations to treat severe forms of hypercholesterolaemia or mixed dyslipidaemia, the focus in these situations is to try to normalize the lipid profile as much as possible while also paying strict attention to patient safety. The clinician should also keep in mind that as the number of drugs to treat dyslipidaemia increases, risk for adverse events increases and drug compliance may decrease. In these situations, it is critical that patients understand warning signs and symptoms for toxicity, and they remain compliant with pharmacological recommendations in order to balance safety with therapeutic success. The use of drugs should always be coupled with therapeutic lifestyle change.


 留言列表
留言列表
 線上藥物查詢
線上藥物查詢 