

Author Information
Adis, a Wolters Kluwer Business, Auckland, New Zealand
Correspondence: Karly P. Garnock-Jones, Adis, a Wolters Kluwer Business, 41 Centorian Drive, Private Bag 65901, Mairangi Bay, North Shore 0754, Auckland, New Zealand.
Abstract
Ecallantide, a recombinant protein that is a selective, highly potent and reversible inhibitor of human plasma kallikrein, is indicated for the treatment of acute attacks of hereditary angioedema (HAE) in patients aged ≥16 years.
In the randomized, double-blind, placebo-controlled, multicentre, phase III trial EDEMA3, mean symptom response to treatment at 4 hours (assessed using the Treatment Outcome Score [TOS]; primary endpoint) was significantly greater with a single subcutaneous dose of ecallantide 30 mg than with placebo in patients with acute, moderate to severe attacks of HAE.
In addition, the mean change from baseline in symptom severity at 4 hours (assessed using the Mean Symptom Complex Severity [MSCS] scale) was significantly greater with ecallantide than with placebo.
At 4 hours in the similarly designed EDEMA4 trial, the mean change from baseline in MSCS score (primary endpoint) and mean TOS were both significantly greater in recipients of a single subcutaneous dose of ecallantide 30 mg than in placebo recipients.
Subcutaneous ecallantide 30 mg was generally well tolerated in patients with acute attacks of HAE in the EDEMA3 and EDEMA4 trials. Adverse events were mostly of mild to moderate severity, and no event that was more common in ecallantide than placebo recipients occurred in >10% of patients.
Hereditary angioedema (HAE) is an autosomal-dominant disorder associated with a C1 esterase inhibitor protein (C1-INH) deficiency.[1,2] It is characterized by swelling as a result of oedema in the interstitium of the deep dermis and subcutaneous tissue, most commonly in the face, gastrointestinal tract, extremities and genitalia, and can affect the laryngeal area to such a degree that the airway becomes compromised.[2] Attacks normally last 2–5 days,[1,2] and patients have an average of one to three attacks per month.[1]
Attacks have been associated with 15 000–20 000 emergency department visits in the US annually, and account for 20–100 sick days per year.[1] The US prevalence of HAE is estimated at 1 :: 10 000 to 1 :: 50 000; sex and ethnic grouping appear to have no effect on the prevalence, but women often have more severe clinical manifestations than men.[1]
There are two main disease types: type I (affects ≈85% of patients and is associated with low serum levels of C1-INH) and type II (affects ≈15% of patients and is associated with normal or elevated levels of C1-INH, but it is dysfunctional).[1,2] A third type of HAE that is not associated with C1-INH deficiency has also been identified.[1,2]
HAE occurs as a result of inappropriate activation of the plasma bradykinin-forming cascade, involving activation of factor XII, which converts prekallikrein to plasma kallikrein, which in turn digests high-molecular-weight kininogen to form the vascular mediator
bradykinin.[2] When present and active, C1-INH inhibits the latter two of these steps, among other roles; a deficiency in functional C1-INH leads to increased bradykinin levels, which leads to increased binding to the bradykinin 2 receptor, associated with oedema and pain.[2,3]
Until recently, treatment options for HAE in the US were limited to short- or long-term prophylaxis with anabolic androgens or oral plasmin inhibitors, which, while effective, can cause dose-dependent adverse events.[4] The recent approval in the US of prophylactic and acute treatment with purified C1-INH (long available in Europe) has added options; however, C1-INH is administered intravenously, and has the potential to be contaminated (e.g. with viral infection).[4] Thus, drug development has focused on agents targeting the kinin cascade downstream of C1-INH.
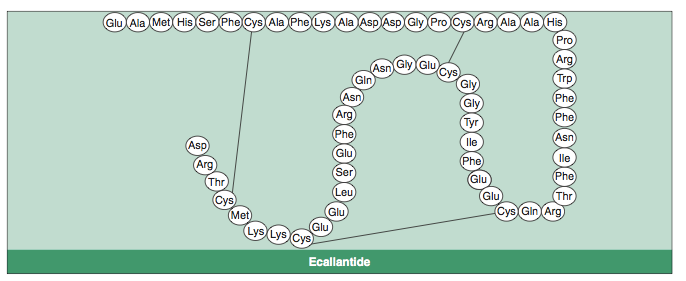
Ecallantide (DX-88; Kalbitor®) is one of these drugs. It is a recombinant protein inhibitor of plasma kallikrein, which is produced in Pichia pastoris, thus escaping contamination risks, and is administered subcutaneously.[4] It is indicated for the treatment of acute attacks of HAE in patients aged ≥16 years.[3] This article provides an overview of the pharmacological properties of subcutaneous ecallantide and reviews the clinical trial data available on the efficacy and tolerability of the drug in this indication.
Medical literature on the use of ecallantide in patients with HAE was identified using MEDLINE and EMBASE, supplemented by AdisBase (a proprietary database). Additional references were identified from the reference lists of published articles.
剩下的文章就摘錄了:
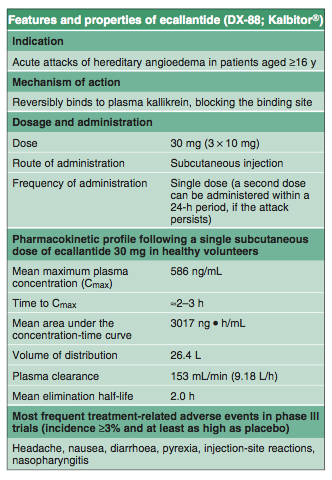
Dosage and Administration
Ecallantide should be administered at a dose of 30 mg as three subcutaneous injections of 10 mg (in 1 mL) in the abdomen, thigh or upper arm, with injection sites separated by at least 5 cm (2 inches) and away from the anatomical site of attack.[3] A second dose can be administered within a 24-hour period, if the attack persists.[3]
The US manufacturer's prescribing information has a black box warning regarding reports of anaphylaxis following ecallantide treatment (section 4).[3] The drug should be administered by a healthcare professional who is familiar with both anaphylaxis and HAE.[3] Patients should undergo observation following ecallantide administration, and it should not be given to patients with known hypersensitivity to ecallantide.[3]
The effectiveness and safety of ecallantide have not been established in patients aged <16 years, and ecallantide is not indicated in these patients.[3] Data are also lacking in patients aged ≥65 years.[3]
Local prescribing information should be consulted for contraindications, precautions and warnings, drug interactions, dosage modifications and patient monitoring requirements.
FDA曾于2008年10月13日批准Cinryze(壹種來源于血漿的C1酯酶抑制劑)用于治療HAE,ecallantide是FDA批准上市的第2只HAE治療藥物,由于該藥與第1只上市新藥相比,具有經皮給藥的特性。


 留言列表
留言列表
 線上藥物查詢
線上藥物查詢 