

N Engl J Med 2014; 370:1164-1166March 20, 2014DOI: 10.1056/NEJMcibr1315508
Heart failure remains a huge health care burden, with one in nine death certificates in the United States listing it as the cause of death.1 Medical therapy has improved the prognosis for patients with heart failure considerably, although there has also been a longstanding dilemma. An ideal heart-failure treatment should improve cardiac function (in the short and long term) and symptoms as well as the prognosis. Unfortunately, current heart-failure drugs that improve the prognosis (e.g., angiotensin-converting–enzyme inhibitors and beta-blockers) do not improve function and symptoms initially (beta-blockers may even worsen them initially), and drugs that improve function and symptoms (e.g., catecholamines and phosphodiesterase inhibitors) actually worsen the long-term prognosis. The notable exception to this rule is cardiac-resynchronization therapy (CRT), in which biventricular stimulation reestablishes coordinated contraction in patients with ventricular dyssynchrony due to left bundle-branch block. Trials have shown that CRT is the only heart-failure treatment that rapidly improves cardiac function and symptoms and reduces long-term mortality.2 What has been less clear is why CRT is so successful, although it is likely that the beneficial mechanisms at work reach beyond reestablishing synchronous contraction.
The contractile state of cardiomyocytes is principally determined by the concentration of Ca2+ ions surrounding the myofilaments; this is sensed by troponin, which is bound to the thin (actin) filaments and serves to regulate cross-bridge cycling. Important posttranslational modifications have been identified as modulating this process: phosphorylation of specific residues of cardiac troponin I, cardiac troponin T, myosin regulatory light chain 2, and cardiac myosin-binding protein C has been shown to modulate the sensitivity of myofilaments to Ca2+ (Figure 1
FIGURE 1
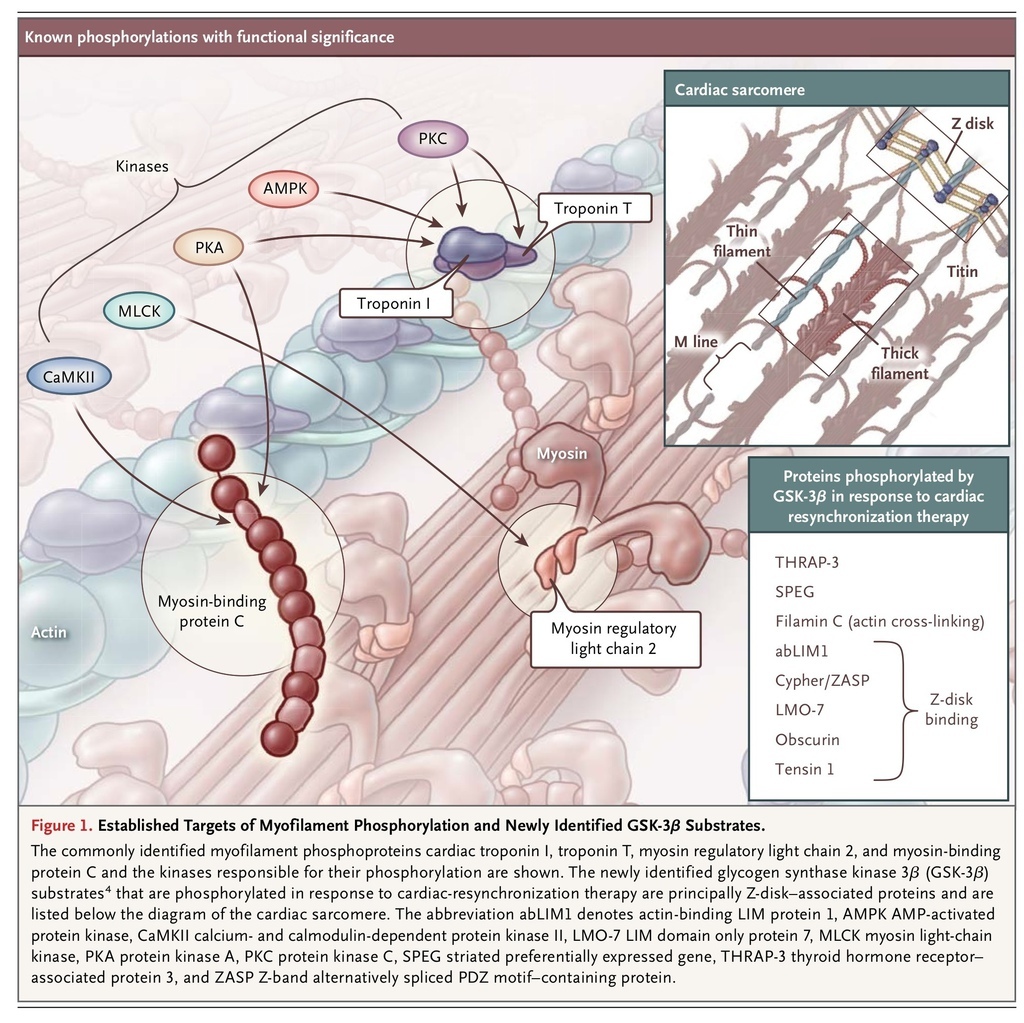
Established Targets of Myofilament Phosphorylation and Newly Identified GSK-3β Substrates.
). Heart failure is typically accompanied by changes in both phosphorylation status and myofilament Ca2+ sensitivity, which are likely to contribute to disease progression.3
Kirk et al.4 recently described strong evidence that the benefits of CRT are related to an improvement in the Ca2+ sensitivity of myofilaments and identified potential molecular pathways involved in this effect. They studied a dog model of heart failure (induced by rapid atrial pacing) and ventricular dyssynchrony (induced by left bundle-branch ablation) and used CRT to reverse the disease. Heart failure was accompanied by decreased myofilament Ca2+ sensitivity, and this was restored by CRT. An important finding was that the benefits of CRT required dyssynchrony to have been present and then reversed; in a heart-failure model of synchronous contraction (rapid atrial pacing without left bundle-branch ablation), Ca2+ sensitivity was as impaired as it was in the dyssynchronous model before CRT.
The observation by Kirk et al. that protein phosphatase 1 treatment decreased myofilament Ca2+ sensitivity in hearts treated with CRT and in control hearts but had no effect on dyssynchronous heart failure strongly suggested that CRT improved myofilament function through phosphorylation. It is intriguing that CRT did not change the phosphorylation status of the commonly identified myofilament phosphoproteins cardiac troponin I, cardiac troponin T, myosin regulatory light chain 2, and cardiac myosin-binding protein C. Mass spectrometry was used to investigate the myofibrillar phosphoproteome, and 13 proteins with increased phosphorylation induced by CRT were identified. Analysis of archival sequence data suggested that the sites were predominantly phosphorylated by glycogen synthase kinase 3β (GSK-3β), a key component of the insulin–phosphoinositide 3-kinase–Akt pathway,5 which is activated in CRT. Further analysis showed that the phosphorylation of sites on eight different proteins, most of which were Z-disk–associated, was both increased by CRT and modified in vitro by GSK-3β (Figure 1). In situ GSK-3β treatment of trabeculae increased myofilament Ca2+ sensitivity in dyssynchronous heart failure but had no effect on control trabeculae or on trabeculae previously treated by CRT, an observation that further implicates phosphorylation by GSK-3β in CRT-mediated recovery of myofilament function. Thus, Kirk et al.4 have provided strong evidence for recovery of myofilament function by CRT through reactivation of GSK-3β.
The study by Kirk et al. adds to our understanding of the effects of CRT in treating heart failure. First, it provides some details of the molecular fingerprint of the benefits of CRT, although to further dissect the signaling pathways involved, it will be necessary to apply the methods that Kirk et al. used in analyzing dyssynchrony and CRT to the mouse heart, with subsequent knockout or overexpression of GSK-3β. Second, their findings lend weight to the concept that myofilament Ca2+ sensitizers are candidate drugs for heart-failure treatment. Such agents are currently in development, although none have been approved for clinical use. Third, GSK-3β is a promising target for heart-failure therapy, and small-molecule activators of this protein might become a new form of treatment. Finally, the observation that a period of preceding dyssynchrony appeared to be necessary for CRT to have a beneficial effect on Ca2+ sensitivity is of interest. The authors suggested that patients with synchronous heart failure might benefit from a period of dyssynchrony (e.g., induced by right ventricular pacing) and that patients undergoing long-term CRT might benefit from intervals of suspended treatment. Experimental studies might test whether such an approach could be effective and, if it is, might determine the optimal timing (duration and frequency) of intermittent periods of dyssynchrony. It is likely, however, that such an approach would not be clinically acceptable because of the risk of decompensation during periods of induced dyssynchrony. Another limitation of the study by Kirk et al. is that the findings were obtained in a specific animal model of heart failure induced by rapid pacing and dyssynchrony induced by ablation. Humans are much more heterogeneous, present with a large number of confounding conditions, and most frequently have heart failure caused by ischemic heart disease or cardiomyopathy rather than induced by tachycardia. Whether the current observations extend to these more common clinical situations remains to be determined.



 留言列表
留言列表
 線上藥物查詢
線上藥物查詢 